|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
ВОСПАЛИТЕЛЬНЫЕ МИОПАТИИ
Воспалительные миопатии представляют собой гетерогенную группу заболеваний, при которых мышечная слабость связана с воспалением мышц инфекционного или аутоиммунного характера. К воспалительным миопатиям относят полимиозит, дерматомиозит, миозит с внутриклеточными включениями, гранулематозный миозит, миозит при вирусных, бактериальных и паразитарных инфекциях, миопатию при саркоидозе и некоторые другие формы. Дерматомиозит является системной иммунозависимой ангиопатией, при которой наблюдаются сосудистые окклюзии и инфаркты, приводящие к развитию всех характерных патологических изменений в мышцах, соединительной ткани, коже, желудочно-кишечном тракте и нервных волокнах. Патогенез связан с образованием антител и иммунных комплексов и активацией системы комплемента. В состав периваскулярного инфильтрата входят Т-лимфоциты, которые в подавляющей своей части являются Т-хелперами, В-лимфоциты и плазматические клетки. Клиническая картина. Пик заболеваемости приходится на возраст 5-10 лет, но описаны случаи и более раннего начала (до 4-месячного возраста). Симптомы возникают постепенно или молниеносно. Скрытое начало характеризуется лихорадкой, недомоганием и утратой аппетита. Мышечная слабость в это время может отсутствовать. Такие неспецифические симптомы сохраняются в течение недель и месяцев, что наводит на мысль о персистирующей инфекции. У большинства детей дерматит появляется раньше миозита. Сыпь вначале локализуется на верхних веках и имеет вид эритемы с очагами нарушенной пигментации и отеком. Затем она распространяется вокруг глаз и на область щек. Эритема и отек на разгибательных поверхностях межфаланговых, локтевых и коленных суставов развиваются позже. Со временем кожа становится атрофичной и шелушащейся. Миопатические изменения включают проксимальную слабость, мышечную ригидность и боль. Слабость нарастает, быстро развиваются сгибательные контрактуры и деформации суставов. Сухожильные рефлексы снижаются, а затем исчезают. У 60% больных обнаруживаются кальцификаты в подкожной клетчатке, особенно под теми областями кожи, где нарушена пигментация. Множественные кальцификаты создают эффект «брони» при рентгенографии. У некоторых детей ведущим начальным симптомом является мышечная ригидность, а кожные и миопатические симптомы выражены не столь ярко. Инфаркты желудочно-кишечного тракта в терминальных стадиях болезни в прошлом приводили к смерти. Летальность при дерматомиозите в настоящее время снизилась и составляет менее 5%, что связано с совершенствованием методов лечения. Более чем у 30% взрослых с дерматомиозитом в дальнейшем выявляются злокачественные новообразования. Диагностика. Сочетание лихорадки, сыпи, миалгии и слабости свидетельствует в пользу диагноза дерматомиозита. В начале болезни уровень КФК обычно повышен. Во время активного дерматомиозита ЭМГ покоя выявляет фибрилляции и положительные острые волны; при мышечном напряжении регистрируются укороченные низкоамплитудные полифазные потенциалы. Мышечная биопсия обнаруживает атрофию миофибрилл. Капиллярные некрозы вначале возникают по периферии мышечного пучка и вызывают ишемию прилежащих миофибрилл. Наиболее выражена атрофия в пучках, которые соприкасаются с большими фасциальными футлярами. Волокна I и II типов (тонические и физические) поражаются в равной степени. Лечение. Воспалительный процесс активен в течение 2 лет. Кортикостероиды снижают его активность, способствуя уменьшению симптоматики. Наилучшие результаты достигаются, когда кортикостероиды назначают на ранних стадиях болезни, в высоких дозах и применяют длительно. Препаратом выбора служит преднизолон. Начальная его доза дается из расчета 2 мг/кг в сутки, но не выше 100 мг/сут. Температура тела часто нормализуется в течение первых 48 час от момента начала терапии. Иногда уровень КФК возвращается к норме на 2-й неделе лечения параллельно с заметным увеличением силы мышечного сокращения. В этом случае дальнейший прием преднизолона может осуществляться по схеме через день и в той дозе, которая позволит снизить тяжесть побочных эффектов стероидной терапии. Терапия преднизолоном одинаково эффективна при приеме препарата ежедневно или по схеме через день, но только в тех случаях, когда лечение не прерывается. Когда сила мышц нарастает, начальную дозу принимаемого через день преднизолона можно снижать на 10% в месяц в течение 5 мес. Дальнейшее уменьшение дозы преднизолона допустимо лишь на 5% в месяц. При решении вопроса о снижении дозы кортикостероидов недопустимо ориентироваться лишь на снижение активности КФК, поскольку заметное увеличение мышечной силы наступает только через 1-2 мес после уменьшения уровня фермента, т.е. ведущим критерием снижения дозы кортикостероидов служит позитивная клиническая динамика. У большинства больных поддерживающая доза преднизолона, принимаемого по схеме через день, которая необходима для нормализации силы мышечного сокращения и концентрации КФК, составляет 25% стартовой дозы. При лечении преднизолоном у некоторых больных сыпь полностью исчезает, но у большинства остаются рубцовые изменения кожи. Длительная стероидная терапия требует контроля функции желудочно-кишечного тракта. Для защиты слизистой оболочки желудка назначают препараты хлорида калия и блокаторов Н2-рецепторов. Серьезным осложнением длительной терапии является развитие стероидной миопатии, которая может быть расценена как обострение основного заболевания. Отличить развивающуюся стероидную миопатию от обострения дерматомиозита по клиническим критериям довольно сложно. При стероидной миопатии, как правило, страдают проксимальные отделы конечностей, развиваются выраженные атрофии, активность КФК не увеличивается. У большинства детей с дерматомиозитом при лечении уже через 3 мес отмечается улучшение, но терапию преднизолоном необходимо продолжать в течение 2 лет. Если лечение прервано преждевременно, неизбежны рецидивы, развиваются кальциноз и контрактуры. Медикаментозное лечение дополняют физической реабилитацией, необходима дыхательная гимнастика. Массаж в активной фазе противопоказан. При правильном лечении благоприятный исход наблюдается у 80% детей с дерматомиозитом. При резистентности или непереносимости преднизолона показано пероральное назначение цитостатиков: метотрексат в дозе от 10 до 20 мг/м2 поверхности тела 2 раза в неделю или азатиоприн в дозе 50-150 мг/сут. Во время терапии необходим регулярный контроль за функцией печени и клеточным составом крови. Комбинация кортикостероидов и цитостатиков позволяет избежать длительной терапии преднизолоном в высоких дозах. В случаях, когда прием кортикостероидов ограничен их побочным действием, применяют плазмаферез или курс внутривенных инфузий иммуноглобулина. В неактивной стадии обострений обычно не возникает. Полимиозит. Этиология в большинстве случаев остается неизвестной. Предполагается, что в патогенезе играют роль клеточные и гуморальные механизмы, что подтверждается нередким развитием заболевания на фоне аутоиммунных процессов (системная красная волчанка, узелковый периартериит, ревматоидный артрит, склеродермия), а также хорошим эффектом применения кортикостероидов и иммуносупрессоров. Таким образом считается, что патогенез связан с клеточно-опосредованной цитотоксической реакцией, реализуемой Т-лимфоцитами, сенсибилизированными к поверхностным антигенам мышечных волокон. Клиническая картина. Полимиозит обычно возникает в зрелом возрасте (45-55 лет), у детей и подростков встречается редко и не связан со злокачественными новообразованиями. Постепенно, исподволь нарастает симметричная проксимальная мышечная слабость, лихорадка и миалгии нетипичны. Часто развивается слабость сгибателей шеи («свисающая голова»). Для заболевания характерны дисфагия и приступы удушья. Постепенно слабость распространяется и на дистальные отделы конечностей. Выраженность парезов варьирует, а в тяжелых случаях развивается тетраплегия. Изредка слабость ограничивается дистальными группами мышц, мышцами глаза или лица. У больного могут наблюдаться периоды стабилизации и даже ремиссии, что может приводить к ошибочной диагностике конечностно- поясной миодистрофии. При хроническом течении болезни постепенно нарастают мышечные атрофии; возможно формирование контрактур. Глубокие рефлексы вызываются на ранних стадиях болезни и снижаются при уменьшении мышечной массы, но полностью никогда не исчезают. Этот важнейший дифференциально-диагностический признак позволяет исключить полиневропатию. Иногда заболевание начинается остро с общего недомогания; резкая мышечная слабость развивается в течение нескольких дней, появляются боли в мышцах плечевого пояса. Атрофии мышц очень легкие или отсутствуют. Часто при рентгенографии в мышцах обнаруживают кальцификаты. У взрослых типичны сердечно-легочные осложнения, нехарактерные для детской формы болезни. Диагностика. Изменения КФК редки. ЭМГ-исследование практически всегда выявляет типичные признаки и миопатического и неврогенного процессов. Мышечная биопсия обнаруживает различные патологические отклонения. Гистологически периваскулярная воспалительная инфильтрация наблюдается не всегда, поэтому отсутствие клеточных инфильтратов в образцах биоптатов не исключает диагноз полимиозита. Для лечения полимиозита используется такая же схема, как при дерматомиозите. Больным, которые резистентны к кортикостероидам, показаны цитостатики (метотрексат). Плазмаферез и внутривенные введения иммуноглобулина являются альтернативными методами при недостаточной эффективности общепринятой терапии. Острый инфекционный миозит возникает после перенесенного гриппа или другой респираторно-вирусной инфекции. Симптомы вирусной инфекции сохраняются от 1 до 7 дней, а затем появляется интенсивная симметричная боль и слабость в мышцах. В тяжелых случаях за 1 сутки больной становится обездвиженным. На фоне общей слабости проксимальные мышечные группы поражены тяжелее, чем дистальные. Болезненна пальпация мышц. Сухожильные рефлексы сохраняются. Уровень КФК обычно более чем в 10 раз превышает верхнюю границу нормы. Почти сразу же вслед за развитием миозита наблюдается его спонтанное обратное развитие. В худшем варианте для исчезновения болевого синдрома требуется от 2 до 7 дней постельного режима, после чего больной полностью выздоравливает. Миозит с включениями. С амая частая миопатия воспалительного характера у взрослых. Ранее это заболевание, в одинаковой степени поражающее и проксимальную, и дистальную мускулатуру, называли полимиозитом, резистентным к глюкокортикоидам. Происходит мутация гена расположенного в 9-й хромосоме. Описаны спорадические и наследственные формы с аутосомно-рецессивным типом наследования. Продолжается дискуссия о том, можно ли считать это состояние первичным дегенеративным заболеванием мышц (в биоптате часто обнаруживают амилоид) или это первичный иммунопатологический процесс (на что указывают воспалительные изменения, выявляемые гистологически –лимфоцитарная инфильтрация, различные размеры мышечных волокон, наличие регенерирующих миоцитов и увеличение объема соединительной ткани). Патоморфологическая картина аналогична таковой при полимиозите, однако с помощью электронной микроскопии в мышце выявляются включения (например, «очерченные» вакуоли, микротубулярные циоплазматические элементы). Дополнительным доказательством дегенератиной природы заболевания стала неэффективность противовоспалительной терапии. Мужчины болеют чаще женщин. Болезнь характеризуется постепенным развитием слабости проксимальной мускулатуры верхних и нижних конечностей у пациентов старше 50 лет, как правило, у мужчин. Чаще всего наблюдается слабость четырехглавой мышцы бедра и сгибателей пальцев, а также дисфагия; однако может формироваться любой другой вариант мышечной слабости. Могут развиться значительная мышечная атрофия, гипо-или арефлексия глубоких рефлексов с конечностей. Не возникает птоза и диплопии, фасцикуляций и нарушений чувствительности и функции тазовых органов. Уровень КФК повышен в 50% случаев. При ЭМГ- и ЭНМГ-исследовании выявляется сочетание миопатических и невральных проявлений, что связано с разрушением мышечной ткани и терминальным аксональным повреждением. Большинство пациентов в течение 10 лет становятся тяжелыми инвалидами, нуждающихся в постороннем уходе. Часто встречаются типичные осложнения обездвиженности: пролежни, тромбоз глубоких вен, аспирационная пневмония и хронические боли при контрактурах суставов. Диагноз ставится на основании результатов биопсии мышцы. Лечение не разработано. Дисфагия указывает на неблаприятный прогноз, может потребоваться наложение гастростомы. Миозит с включениями необходимо заподозрить во всех случаях предполагаемого полимиозита, в случае, если не эффективны кортикостероиды.
АМИОТРОФИИ
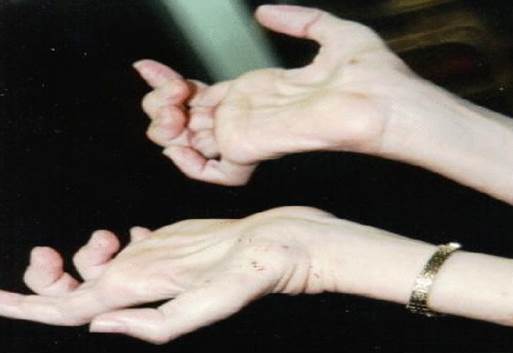
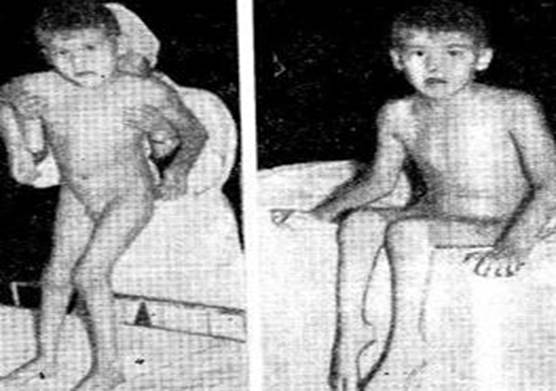
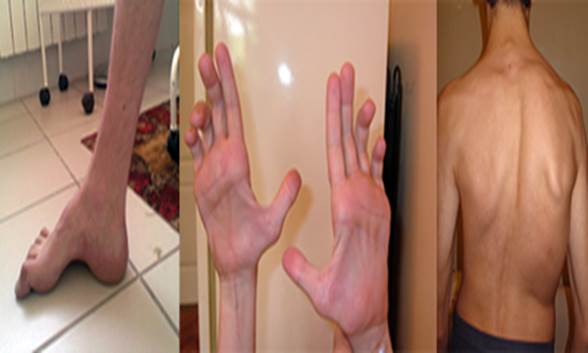
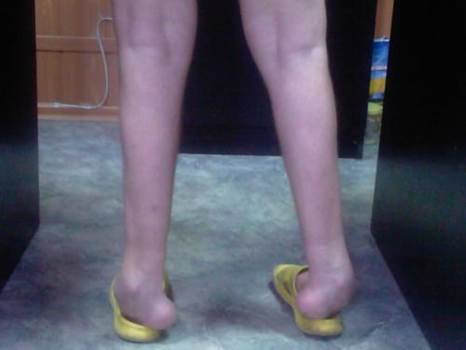
Амиотрофии - группа заболеваний, обусловленных поражением мотонейронов спинного мозга, периферических нервов и двигательных клеток мозгового ствола, вызывающих атрофию и слабость мышц. Они характеризуются постепенным развитием параличей, качественной реакцией перерождения соответствующих мышц, снижением их электровозбудимости. Атрофии подвергаются как саркоплазма, так и миофибриллы. Развивается денервационная, вторичная атрофия мышечного волокна в результате нарушения его иннервации, в отличие от первичного атрофического процесса в мышцах. Выделяют спинальные и невральные амиотрофии. Невральные амиотрофии -наследственные формыполиневропатии (гетерогенных генетически и электрофизиологически). В настоящее время установлено 7 типовнаследственных мотосенсорныхневропатий (НМСН). На основании электрофизиологических критериев выделяют две группы: тип 1(демиелинизирующие НМСН) итип 2 (аксональные НМСН), которые представляют собой варианты болезни Шарко-Мари -Тута. Болеют лица обоего пола. НМСН1 типа встречается с частотой 1: 50000 населения, чаще аутосомно- доминантный тип наследования, значительно реже - аутосомно-рецессивный или рецессивный, сцепленный с Х-хромосомой. Возможны и спорадические случаи. Происходят мутации 17-й хромосомы (17g11.2-12) или 1-й хромосомы(1 g-22-23), в результате нарушается синтез миелина с развитием демиелинизирующейполиневропатии. НМСН I типа составляет 60% всех случаев болезни Шарко-Мари. Патоморфология: сегментарнаядемиелинизация впериферических нервах, наряду с этим выявляются утолщения по типу «луковичных головок», отражающие процессы ремиелинизации. В мышцах возникают денервационные изменения с явлениями «пучковой» атрофии мышечных волокон. Клиника. Первые признакиболезни возникают в возрасте 15-30 лет, реже в более молодом возрасте. В начале болезнивозникает мышечная слабость, патологическая утомляемость, преимущественно в дистальных отделах конечностей. Примерно у половины больных при пальпации выявляется утолщение нервных стволов. НМСН 2 типа ( аксональная форма) – первично поражаются аксоны двигательных волокон периферических нервов. Наследуется по аутосомно- доминантному или аутосомно-рецессивному типу.Мутации могут происходить в генах 1,8,3,7-й хромосом. Один из установленных биохимических дефектов связан с геном, кодирующим легкие цепи нейрофиламентов (белки цитоскелетамотонейронов). Патоморфология:Обнаруживается гибель аксонов периферических двигательных нервов и вторичная сегментарнаядемиелинизация без утолщений по типу «луковичных головок», так как процессы ремиелинизации отсутствуют. Клиникасходна с НМСН 1 типа, однако возникает в более позднем возрасте часто на 2-ом десятилетии жизни, в отдельных случаях на 6-7 десятилетии жизни; мускулатура рук реже вовлекается в патологический процесс, менее выражены чувствительные нарушения. Заболевание медленно нарастает. ЭНМГ – отсутствует существенное снижение скорости проведения по нервам. В клинической картине вначале заболевания появляется слабость в ногах, особенно разгибателя большого пальца и передней большеберцовой мышцы. Вскоре выявляется атрофия этих мышц и мелких мышц стопы; стопа начинает отвисать, вследствие чего походка нередко приобретает характер петушиной (степпаж от англ. steppere - трудовая лошадь). Избирательная атрофия мышц дистальных отделов нижних конечностей придает им своеобразную форму, которую сравнивают с перевернутыми бутылками или ногами аиста. Атрофия мышц стопы приводит к когтевидной установке пальцев и деформации стоп (свод их очень высок), напоминающие стопу Фридрейха; положение стоп становится неправильным (pes varus, pes equinus). Уже в ранних стадиях болезни исчезают ахилловы рефлексы. Нередко больные жалуются на парестезии и стреляющие боли в ногах. Выявляется расстройство чувствительности по полиневральному типу, причем поверхностные виды чувствительности (главным образом, болевая и температурная), страдают в значительно большей степени. Могут возникать боли в конечностях, повышенная чувствительность к давлению нервных стволов. В дистальных отделах конечностей наблюдаются и вазомоторно-трофические нарушения (цианоз, отек, краснота, расстройство потоотделения). Атрофический парез нарастает медленно; обычно через 5—10 лет в процесс вовлекаются и верхние конечности: сначала атрофируются мелкие мышцы кистей, а затем и предплечий. Кисти приобретают форму "обезьяньей лапы". Исчезают глубокие рефлексы с конечностей.Часто наблюдаютсянерезко выраженные фасцикулярные подергивания в мышцах конечностей. Мышцы тазового и плечевого пояса, проксимальных отделов конечностей, туловища, шеи, лица, как правило, не страдают, поэтому больные длительное время сохраняют возможность самообслуживания. Клинические проявления при невральной амиотрофии Шарко- Мари-Тута могут варьировать в отношении времени и последовательности появления, степени выраженности основных симптомов. Тяжесть двигательных расстройств, развивающихся при этом, может быть различной. Иногда отмечаются симптомы поражения передних рогов спинного мозга (фибриллярные подергивания в мышцах), атрофия зрительных нервов, мозжечковые симптомы, постуральный тремор в руках и др. Течение медленно прогрессирующее. Иногда процесс обостряется в связи с различными экзогенными вредностями. В отдельных случаях в течение длительного времени состояние больных может оставаться стационарным. Нередко больные доживают до глубокой старости, сохраняя возможность самообслуживания и даже трудоспособность. При НМСН I типа целесообразна иммунотропная терапия (кортикостероиды, иммуноглобулин, плазмаферез). При ЭНМГ выявляется симметричное равномерное замедление скорости проведения возбуждения по всем исследуемым нервам – как правило к 38 м/. Лечениеневральной амиотрофии направлено на прекращение дегенерации нервов. К сожалению, этот процесс нельзя повернуть вспять, можно только остановить. Поэтому важно своевременно начать лечение. Проводится симптоматическая терапия: назначают антихолинэстеразные препараты, витамины группы В и Е, высокие дозы аскорбиновой кислоты, анаболические гормоны, биостимуляторы, АТФ, глутаминовую кислоту, коэнзим а, аутогемотерапию, массаж, ЛФК, физиотерапию (электрофорез и ультразвук) и бальнеотерапию (радоновые исероводородные ванны).Лечение следует проводить повторными курсами. При свисающих стопах показана ортопедическая помощь (специальная обувь, в тяжелых случаях - тенотомия). Существенную роль имеет правильный выбор профессии, не связанной с большим физическим утомлением. Профилактиканевральной амиотрофии заключается в медико-генетическом консультировании. Интерстициальная гипертрофическая невропатия Дежерина- Сотта встречается редко. Принадлежность заболевания к невральным амиотрофиям не доказана. Клинически оно сходно с невральной амиотрофией Шарко-Мари-Тута, однако заболевание начинается в раннем детском возрасте. Отличительной чертой является также утолщение нервных стволов (гипертрофический неврит) в результате разрастания в них соединительной ткани и гипертрофии шванновских клеток. Спинальные амиотрофии обусловлены поражением мотонейронов поясничного и шейного утолщений спинного мозга. Спинальная амиотрофия Верднига-Гоффманна (син. прогрессирующая спинальная амиотрофией детского возраста, злокачественная инфантильная спинальная амиотрофия, спинальная мышечная атрофия (СМА)) проявляется прогрессирующей постепенно нарастающей слабостью и атрофией мышц туловища и проксимальных отделов нижних и верхних конечностей. Часто эта амиотрофия возникает при кровном родстве между родителями. Аутосомно-рецессивный тип наследования с мутацией гена на 5-й хромосоме (5 g 11.2-13.3), в результате нарушается синтез белка, поддерживающего жизнеспособность мотонейрона (motor neuron survival protein), предположительно участвующего в синтезе РНК. Частота заболевания –7 случаев на 100000 новорожденных Патоморфология.Отмечается дегенерация спинальных мотонейронов и передних корешков,часто – и в двигательных ядрах и корешках Y, Y1, YП,1Х, Х, Х1 и ХП черепных нервов. В скелетных мышцах вторичные нейродегенеративные изменения (пучковая атрофия и др). Выделяют врожденную форму, раннюю детскую форму и позднюю форму этого заболевания. Врожденная форма (СМА I типа). В 13 случаев клиническая картина развертывается внутриутробно (вялое шевеление плода), в других случаях симптомы болезни возникают в первые 3 месяца жизни младенца. В большинстве случаев наблюдаются вялые парезы мышц проксимальных конечностей, мышечная гипотония, снижение или утрата глубоких рефлексов. Возникают бульбарные расстройства (вялое сосание, слабый крик, фасциркуляции в мышцах языка, снижение глоточного рефлекса.). Развитие статических и моторных функций резко замедлено. Характерны костно-суставные деформации (сколиоз, воронкообразная или «куриная» грудная клетка, контрактуры суставов). Часто выявляются врождённые пороки развития (гидроцефалия, крипторхизм, гемангиома, дисплазия тазобедренных суставов, косолапость). Заболевание быстро прогрессирует, главными причинами смерти больных детишек является нарастающая лёгочная и сердечно-сосудистая недостаточность из-за слабости дыхательной мускулатуры и соответствующих изменений в корковых отделах головного мозга. Ранняя детская форма (СМА II типа) может возникать в период от полугода до года после рождения. Сначала поражаются мышцы ног и туловища, затем патологический процесс захватывает все группы мышц, включая мышцы, иннервирующиеся черепными нервами. Ребёнок не может опираться на ножки, не способен сидеть и хватать игрушки. В сидячей позе развивается сильно выраженный кифоз по причине слабости мышц спины. Рано выпадают глубокие рефлексы с конечностей. Часто отмечаются фасциркулярные подёргивания мышц, особенно мышц языка. В поздней фазе болезни наблюдается полная мышечная гипотония и бульбарный паралич. Продолжительность жизни не более 14-15 лет. Для поздней формы амиотрофии Верднига-Гоффмана характерно постепенное начало в возрасте 1,5-2,5 лет. В походке и движениях ребёнка наблюдается нарастающая неуверенность, дети начинают часто падать. В дальнейшем, к этому присоединяются вялые параличи и атрофии проксимальных отделов нижних и верхних конечностей. Снижаются или полностью выпадают глубокие рефлексы, развивается кифосколиоз и контрактуры. Возникает бульбарный паралич с атрофией и фасциркуляциями в мышцах языка, снижением глоточного и нёбного рефлексов, дисфагией. Двигательные навыки начинают медленно исчезать, и к 10-12 годам дети становятся обездвиженными. Доживают такие больные при хорошем уходе до 20-30 лет. Диагноз.На ЭМГ при спинальной амиотрофии Верднига-Гоффманна отмечается спонтанная биоэлектрическая активность в покое с наличием потенциалов фасцикуляций. При произвольных сокращениях регистрируется уреженная электрическая активность с ритмом «частокола». Активность ферментов сыворотки крови не меняется. Лечение при спинальной амиотрофии Верднига - Гоффманна сводится к назначению массажа и ЛФК, которые должны проводиться систематически. Радикального лечения не имеется.Некоторое улучшение оказывают такие препараты, как церебролизин, аминалон, элькар, антихолинэстеразные средства, витамины группы В, повторное (4-5 раз) переливание малых доз одногруппной крови по 50 мл. Прогноз неблагоприятный. Известен особый вариант спинальной атрофии Верднига-Гоффманна. П рогрессирующий бульварный паралич или болезнь Фацио-Лонде. Заболевание чаще начинается к концу второго года жизни, иногда в ювенильном возрасте, характеризуется слабостью в мышцах лица, включая жевательную мускулатуру, появляются дисфагия, дисфония, атрофии в мышцах языка. Может наблюдаться офтальмоплегия. Заболевание быстро нарастает, летальный исход наступает спустя 6-12 мес. от появления первых симптомов. К бульбарным расстройствам могут присоединяться вялые парезы верхних и нижних конечностей, иногда клинически они не успевают развиться, однако на вскрытии постоянно констатируется поражение мотонейронов спинного мозга на всем протяжении. Описаны семейные случаи болезни Фацио - Лонде [Alexander et al., 1976], когда страдали два и более сибса. Тип наследственной передачи — аутосомно-рецессивный. Дифференциальный диагноз спинальной амиотрофии проводят с: Острый полиомиелит - для него характерно острое начало с лихорадкой, параличи не симметричны и отсутствует прогрессирование процесса. Миопатии - тоже прогрессирующее наследственное заболевание мышц, но связано оно с нарушением обмена веществ в мышечной ткани. При миопатиях нет подергиваний мышц, не такое быстрое течение и другая электромиографическая картина. Врожденная миатония (нарушение тонуса мышц) - вот от нее спинальную миотрофию отличить не так просто. Основной признак врожденной миатонии - распространенная и сильно выраженная гипотония (снижение тонуса) мышц. Миаотния отличается от спинальной амиотрофии доброкачественным течением и отсутствием подергиваний мышц. Отличить эти два заболевания друг от друга помогает биопсия (взятие образца ткани) мышц. Псевдомиопатическая (юношеская) форма спинальной амиотрофии Кугельберга — Веландера. В 1942 г. Wohlfart впервые описал заболевание, проявляющееся мышечными атрофиями ипарезами, напоминающее миопатию, но с распространенными фасцикуляциями. В 1956 г. Kugelberg и Welander подчеркнули, что такое заболевание протекает сравнительно доброкачественно; тщательный электромиографический контроль позволил авторам уточнить неврогенный характер мышечной атрофии и классифицировать последнюю как спинальное поражение. Частота не установлена. Тип наследственной передачи — аутосомно-рецессвный, реже - аутосомно- доминантный, рецессивный сцепленный с Х-хромосомой. Генетический дефицит – в том же локусе, что и при других спинальных амиотрофиях. Заболевание начинается обычно в возрасте 4-8 лет, иногда позднее. Появляются общая слабость, повышенная утомляемость, слабость в ногах (особенно при подъеме по лестнице), подергивания в мышцах. Развиваются вялые парезы нижних конечностей с фасцикуляциями. Нередко у больных отмечается избыточное развитие подкожно-жировой клетчатки, маскирующей развитие атрофии и подергивания мышц. Изменяется походка, снижается тонус мышц, исчезают глубокие рефлексы с нижних конечностей. По клиническим симптомам заболевание напоминает конечностно-поясную миопатию Эрба-Рота. При осмотре также отмечается псевдогипертрофия икроножных мышц (увеличение их в объеме за счет развития жировой ткани) Через несколько лет после начала заболевания появляются атрофии и фасцикулярные подергивания в мышцах проксимальных отделов верхних конечностей. Течение медленно прогрессирующее, двигательная активность сохраняется длительно. Больные живут до 40—50 лет, нередко сохраняя возможность самообслуживания. При появлении в поздних стадиях бульбарных симптомов прогноз ухудшается. Дополнительные исследования при спинальной амиотрофии Кугельберга - Веландера выявляют довольно своеобразные изменения - электромиография указывает на отчетливые признаки спинального поражения, в то же время патоморфологическая картина при биопсии мышц представлена смешанным характером патологии - наряду с неврогенной амиотрофией имеются указания и на некоторые дистрофические признаки. Аналогичные данные получают и при биохимическом исследовании - активность ферментов, в том числе креатинфосфокиназы нередко повышена, хотя и в меньшей степени, чем при истинной миопатии. Изменяются показатели креатин-креатининового обмена. Специфического лечения при амиотрофии Кугельберга - Веландера нет. Применяются симптоматические и общеукрепляющие средства. Важное значение имеет правильный выбор профессии, устранение физических перегрузок. Болезнь Арана-Дюшена (спинальная амиотрофия взрослых) начинается в возрасте 40-60 лет. Постепенно возникает нарастающая атрофия мышц дистальных отделов конечностей (преимущественно кистей). В дальнейшем в процесс могут вовлекаться и мышцы проксимальных отделов верхних и нижних конечностей. В пораженных мышцах конечностей и языка отмечаются фасцикулярные подергивания. Течение медленно прогрессирующее. Смерть обычно наступает от бронхопневмонии. Амиотрофия бульбоспинальная поздняя Кеннеди. Впервые заболеваниеописали в 1968г. W.R. Kennedy и соавт. Наследуется по рецессивному, сцепленному с Х хромосомой типу. Причиной является специфическая мутация в гене андрогенового рецептора, картированного в Х-хромосоме. Болеют только мужчины. Манифестирует обычно после 30 лет. Патоморфология.Выявляют дегенерацию и умеренный глиоз части мотонейронов спинного мозга и в двигательных ядрах ствола. Клиника.Заболеваниепроявляется в зрелом возрасте, чаще в 20-40 лет, генерализованными фасцикулярными подергиваниями, умеренной слабостью и атрофией мышц. При этом глубокие рефлексы, как правило, снижены или отсутствуют. Преобладают проксимальные амиотрофии с поражением прежде всего мышц тазового пояса. В последующем медленное распространение патологического процесса, поражение мышц плечевого пояса, языка и глотки с умеренно выраженным бульбарным синдромом, а так же слабость жевательных и мимических мышц. Характерны фасцикуляции в периоральной мускулатуре и языке. Развитие миодистрофического процесса ведет к утомляемости при ходьбе, изменению походки, затруднению при подъеме по лестнице, которые медленно нарастают. Возможны тремор рук, головы, напоминающий эссенциальное дрожание и судороги по типу крампи. Обычны эндокринные расстройства: сахарный диабет, расстройства функций половых желез (гинекомастия, бесплодие). Встречается очень редко. Течение медленное, социальный прогноз в основном благоприятен. Скапуло-перонеальная форма Вюльпиана спинальной амиотрофии начинается в возрасте между 20-40 годами с атрофии мышц плечевого пояса (ограничение движений в плечевых суставах, «крыловидные» лопатки), со временем присоединяется слабость разгибателей стоп, голеней, хотя возможна и обратная последовательность. Прогрессирование медленное, нередко пациенты с 30-40-летним сроком заболевания самостоятельно передвигаются. Дифференциальный диагнозследует прежде всего проводить со скапуло-пероеальной миодистрофией Давиденкова. Лопаточно-перонеальная амиотрофия Происходит мутация гена,картированного в областихромосомы 1q21.2, ответственного за синтез белка Ламин. Передается по аутосомно-доминантному или аутосомно-рецессивному типу. Проявляется проксимальным поражением верхних конечностей и дистальным - нижних. Начинается заболевание сравнительно поздно - в 30-40 лет и старше. Наблюдаются концевые атрофии, например в большой грудной мышце, иногда крупные фасцикулярные подергивания, угнетение сухожильных рефлексов. В ряде случаев слегка нарушается чувствительность - дистальные парестезии, гипестезии, иногда умеренные боли. Отмечается реакция перерождения, обычно умеренно выраженная. При электромиографическом исследовании выявляются специфические изменения, отличающие эту форму от обычной миопатии и невральной атрофии Шарко - Мари (дизритмичные колебания в покое, снижение амплитуды, уменьшение частоты и иногда групповые спайковые разряды при активных движениях). Таким образом, данная форма является как бы промежуточной между первичной миопатией и невральной амиотрофией. Диагноз. Заподозрить спинальную амиотрофию можно при наличии вялых параличей определенной локализации, атрофии мышц с фасцикулярными подергиваниями в них, арефлексией глубоких рефлексов, прогредиентном течении заболевания и др. Учитываются данные дополнительных исследований: -ЭМГ, ЭНМГ. При тотальной ЭМГ признаки переднерогового поражения: в режиме покоя спонтанная биоэлектрическая активность в виде потенциалов фисцикуляций амплитудой до 200 мкв, при произвольном сокращении уреженная ЭМГ с ритмом «частокола» ирезко разряженная при тяжелом поражении мышц. На ЭНМГ — снижение амплитуды М-ответа и числа функционирующих двигательных единиц. Скорость проведения импульса варьирует в зависимости от формы спинальной амиотрофии и возраста больного; биопсия мышц (типична пучковая атрофия в отличие от диффузной при ПМД); -МРТ. Иногда позволяет выявить атрофию спинного мозга; -исследование активности КФК, ЛДГ, АЛТ в сыворотке крови (в отличие от ПМД нормальна или незначительно повышена); биопсия пораженных мышц. Профилактика посредством медико-генетического консультирования.
МИОТОНИИ Миотонии (myotoniae; от греч. myos мышца + tonos напряжение) группа наследственных нервно-мышечных заболеваний, относящихся к группе мышечных каналопатий. Миотония проявляется миотоническим феноменом - тоническим спазмом мышцы после активного напряжения с замедленным расслаблением. Феномен миотонии обусловлен усилением нестабильности мембраны мышечного волокна (сарколеммы). В зависимости от этиологического фактора различают миотонию действия, перкуссионную и электромиографическую. Миотония действия: Важно убедиться в существовании затруднений в начале активных движений, совершающихся с мышечным усилием. При этом, если пациента попросить сжать кулак и быстро его разжать, то расслабление после активного напряжения мышцы затруднено, сократившаяся мышца как бы стремится удержать свое состояние напряжения, фаза расслабления растягивается на 5-30 с. При повторных движениях интенсивность спазма уменьшается, и после 5 -7 повторений движение становится нормальным. Число сокращений, необходимых для устранения спазма, является показателем выраженности заболевания и может иметь значение при оценке трудоспособности больных. Миотония перкуссионнаяпроявляется мышечным сокращением при энергичном ударе неврологическим молоточком, при этом на месте удара образуется углубление («миотонический ровик»), сохраняющееся 4-8 с.; Миотония электромиографическаяпроявляется тем, что при игольчатой ЭМГ активное движение или перкуссия мышц вызывает появление высокочастотных повторных разрядов, которые вначале увеличиваются по частоте и амплитуде, а затем уменьшаются (при прослушивании звуковой эквивалент в виде феномена «пикирующего бомбардировщика»). По классификации D. Gardner-Medwin, Y. Walton (1969) среди наследственных миотоний различают: - врожденную миотонию Томсена; - врожденную миотонию Беккера; - дистрофическую миотонию; - миотонию хондродистрофическую; - врожденную парамиотонию Эйленбурга; - парамиотонию с параличом, возникающим на холоде. Классификация миотонических заболеваний В. С. Лобзина и соавт. (1979): I. Наследственные формы миотоний А. Стационарные медленно прогрессирующие формы: врожденная миотония Томсена; врожденная миотония Беккера; приобретенная миотония Тальма; дистрофическая (атрофическая) миотония Гоффманна - Россолимо - Штейнерта - Куршманна; неонатальная форма дистрофической миотонии. Б. Периодические (рецидивирующие) формы миотонии: интермиттирующая миотония Марциуса -Ганземана; врожденная парамиотония с холодовыми параличами Эйленбурга; врожденная парамиотония без холодовых параличей Де Ионга; эпизодическая наследственная миотоническая адинамия Беккера. II. Миотонические синдромы: -при миопатиях; -при периодическом параличе и эпизодической адинамии Гамсторпа; -при органических болезнях центральной нервной системы; -призаболеваниях внутренних органов; -при синдроме Швартца - Джемпела, нейромиотонии, гипофункции щитовидной железы, начальных стадиях полимиозита и др. Различные формы миотоний отличаются разным типом наследования, вариабельностью возраста проявления заболевания, его течения и другими признаками. К наиболее часто встречающимся формам относятся врожденная миотония и дистрофическая миотония. Врожденная миотония встречается в двух вариантах: миотонии Томсена с аутосомно-доминантным типом наследования и миотонии Беккера с аутосомно-рецессивным типом наследования. Болеют лица обоего пола. Обе формы заболевания имеют схожую клинику, но миотония Беккера характеризуется более поздней манифестацией и более тяжелыми клиническими проявлениями. Врожденная миотониявстречается в Европе с частотой 1: на 100 000 населения, в странах Скандинавии – 1: 100 000 населения, причем миотония Беккера встречается в три раза чаще миотонии Томсена. Патоморфология. Специфические патоморфологические признаки при миотонии отсутствуют. В большинстве случаев выявляется вариабельный диаметр мышечных волокон, их гипертрофия и централизация ядер. Этиология. Формыврожденной миотонии Томсена и Беккера являются аллельными вариантами мутаций в гене СLC№1, картированного в области длинного плеча 7-й хромосомы (7g35)4; в норме он кодирует белок хлорных каналов скелетных мышц. Патогенез. Белок мышечных хлорных каналов регулирует электрическую возбудимость мембраны скелетных мышц. В отсутствии и при функциональной неактивности белка нарушается проникновение хлора в мышечное волокно, что приводит к возникновению электрической нестабильности мембраны. Имеют значение повышенная чувствительность ткани к ацетилхолину и калию. Миотония Томсена описана Thomsen в 1876 г. Заболевание характеризуется тоническим спазмом, возникающим в различных группах мышц в виде затруднения расслабления мышц после активного напряжения. Проявляется уже на 1-м году жизни изменением голоса и медленным расслаблением мышц лица при плаче. Трудность расслабления глоточных мышц вызывает нарушение глотания. Миотоническая реакция в мимических мышцах усиливается при сосании и на холоде. Выраженность спазма наибольшая в начале движения и уменьшается во время повторных мышечных сокращений. Наиболее часто первые признаки заболевания возникают в дистальных отделах рук. По мере прогрессирования заболевания в патологический процесс вовлекаются мышцы ног, а также жевательная и мимическая мускулатура. Пассивные же движения, так же как и слабые медленные активные движения миотоническим спазмом не сопровождаются. Дети, страдающие миотонией, в физических играх уступают сверстникам. Характерны жалобы на затруднения в самом начале активных движений, особенно если движения совершаются быстро и со значительным усилием. Фаза расслабления мышц задерживается на продолжительное время, и больные не в состоянии быстро разжать кисти, изменить положение ног, быстро открыть зажмуренные глаза, быстро встать со стула. Отмечается утомляемость при физических нагрузках. Характерно возникновение перкуссионных миотонических феноменов в виде валика или ямки. Активная миотония в виде трудности расслабления мышц в начале произвольного движения появляется в более позднем возрасте. Ходьба, особенно после длительного покоя, затруднена, первые шаги совершаются с большим усилием. Перед подъемом по лестнице больной «замирает» (время на расслабление мышц), подняться на первую ступеньку для него представляет большую трудность, чем дальнейший подъем. Застывание и скованность, постепенно устраняются после повторных однотипных движений. Болезнь протекает мягко, необычные явления (затруднение расслабления мышц) становятся актуальными в возрасте 12-18 лет (игры, спорт, призыв в армию). Больные имеют атлетическое телосложение, мышцы плечевого пояса, особенно дельтовидные хорошо контурируют. При пальпации мышцы плотные, твердые, однако объективно мышечная сила снижена. Миотонические спазмы возможны в различных группах мышц, но чаще в кистях и ногах, круговых мышцах глаза, жевательной мускулатуре. При охлаждении и волнении может наступить тотальный мышечный спазм во всем теле: больные падают как подкошенные и не могут быстро встать. Сильное сжатие пальцев кисти, длительное статическое напряжение ног, смыкание челюстей, зажмуривание глаз вызывают тонические спазмы. При ударе по мышце неврологическим молоточком при миотонии на месте удара образуется углубление или мышечный валик, сохраняющееся несколько секунд (миотония перкуссионная). Электровозбудимость мышц также нарушена: после раздражения мышцы фарадическим или гальваническим током отмечается замедленность ее расслабления (миотония электромиографическая). При повторных движениях миотонические спазмы постепенно уменьшаются. Повышение механической возбудимости мышц выявляется помощью специальных приемов: при ударе неврологическим молоточком по возвышению I пальца происходит приведение его к кисти (от нескольких секунд до минуты) – «симптом большого пальца», при ударе перкуссионным молоточком по языку на нем появляется ямка – «симптом языка». В мышцах подчас возникают болезненные ощущения, особенно в холодное время года. С возрастом миотонический феномен становится менее выраженным. Глубокие рефлексы с конечностей могут быть нормальными, а в тяжелых случаях снижены. Миотонический феномен на протяжении всего периода заболевания отмечается преимущественно в мышцах лица, глотки, жевательной мускулатуре, верхних конечностей. Усиление миотонического феномена возникает на холоде, уменьшение - в тепле, во время отдыха и при приеме небольших доз алкоголя. Диагностика.У части больных может быть увеличен уровень КФК в плазме крови. На ЭМГ выявляются миотонические феномены: после прекращения активного движения (например, сжимание кисти в кулак) в фазе расслабления обнаруживают биоэлектрическую активность не только в мышцах-разгибателях, но и мышцах-сгибателях (так называемый феномен интерференции, амплитуда которой значительно уменьшается по мере расслабления мышцы). Трудоспособность сохраняется в течение длительного времени. Качество жизни во многом зависит от своевременной и правильной профориентации – рекомендованы специальности, не связанные с быстрыми движениями. Противопоказана работа шофера, летчика, на конвейере и др. Продолжительность жизни не нарушается. Профилактика основывается на возможности дородовой ДНК-диагностики. Миотония Беккера описана Becker в 1977 г. Болеют лица обоего пола. Начинает проявляться у девочек в возрасте от 4 до 12 лет, а у мальчиков - в возрасте 18 лет. Клинические проявления заболевания сходны с миотонией Томсена, но более выражены, у части больных развивается слабость проксимальных групп мышц. Мышечные гипертрофии встречаются редко. У пациентов отмечается неловкость движений. Сначала поражаются мышцы ног, в дальнейшем наступает постепенная генерализация: через несколько лет мышц поражаются мышцы рук, а на поздних стадиях - жевательная и мимическая мускулатура. Прогредиентность выражается также в развитии, спустя 6-8 лет от начала заболевания, слабости и атрофии мышц преимущественно плечевого пояса и проксимальных отделов верхних конечностей. Возникает ригидность мышц. При биопсиимышц на поздней стадии заболевания, помимо миотонических признаков обнаруживают умеренно выраженные миодистрофические изменения. Уровень КФК в плазме крови может быть повышен. Возможна дородовая ДНК-диагностика. Дистрофическая миотония Гоффмана-Россолимо-Штейнерта- Куршманна (DM) впервые описана Г.И. Россолимо (1901), более подробно изложена H. Steinert и F.F. Batten в 1909г. Является мультисистемным заболеванием, при котором отмечается нарушение функционирования различных органов и тканей (в том числе, мышечной, сердечнососудистой, дыхательной, пищеварительной и эндокринной), а также когнитивных функций. Тип наследования аутосомно-доминантный. Является самой частой формой среди миотоний. Клиническая картина складывается из миотонического и миопатического синдромов, а также вегетативно-трофических нарушений. По сути, дистрофическая миотонияявляется переходной формой между миотонией и миопатией. Заболевание представлено тремя подтипами. Так, дистрофическая миотония 1-типа (DM1) возникает вследствие мутации гена DMPK в области длинного плеча 19-й хромосомы (19q13), ответственного за синтез белка миотонин-протеинкиназы, который в норме определяется в скелетной и гладкой мышечной ткани, в миокарде, а также ЦНС, фибробластах, лимфоцитах. В норме количество число повторов тринуклеотида цитозин-тиамин-гуанин (ЦТГ) варьирует от 5 до 37, а при DM1 увеличивается от 38 до нескольких тысяч. При этом ранний дебют и наиболее тяжелое течение заболевания отмечается при числе ЦТГ - повторов равных 3000 и более. Мутация гена DMPK приводит к нарушению гомеостаза ионов Ca++, а также процессов возбуждения и сокращения мышц, сердечной проводимости. DM1 составляет 98 % случаев среди дистрофической миотонии. DM2 возникаетвследствие мутации гена в области длинного плеча 3-й хромосомы (3q21). В то время, как для DM1 характерна мышечная слабость в дистальных отделах конечностей, то для DM2 - в проксимальных. DM3 возникает вследствие мутации гена в области длинного плеча 15-й хромосомы (15q21-q24). Классификация дистрофической миотонии: - конгенитальная (врожденная, congenital myotonic dystrophy — CmyD) - с характерной клинической симптоматикой сразу при рождении ребенка (описана только при DM1); - ювенильная (juvenile DM) - с дебютом заболевания в возрасте у детей в возрасте от 1 года до подросткового периода; - дистрофическая миотония взрослых (adult DM) - с дебютом заболевания у больных в возрасте от 20 до 40 лет; - дистрофическая миотония с поздним дебютом (DM with late onset) - с возникновением заболевания у больных в возрасте старше 40 лет. Эпидемиология. Распространенность DM1 в больших популяциях - около 1: 8000, при этом наиболее часто (1: 3000) встречается в Швеции. Распространенность DM2 и DM3 не досточно изучена. Патоморфология. При патоморфологическом исследовании в скелетных мышцах выявляется атрофия отдельных групп мышц, а в пораженных мышечных волокнах - изменения, свойственные миопатии и миотонии. При дистрофической миотонии нередко наблюдаются бронхоэктазы и эмфизема лёгких, которые связывают с гиповентиляцией и повторными инфекциями. Со стороны эндокринной системы отмечаются атрофия яичек и коры надпочечников, аденомы щитовидной железы. Изменения в нервной системе в одних случаях представлены атрофией двигательных нейронов спинного мозга, гидроцефалией со значительным расширением III желудочка головного мозга, атрофией периферических нервов, в других случаях патологические изменения в нервной системе отсутствуют. Клиника. Различные типы дистрофической миотонии имеют схожую клиническую картину, могут быть дифференцированы с помощью анализа ДНК. Болезнь чаще манифестирует на 3-4-м десятилетиях жизни. Мужчины и женщины болеют с одинаковой частотой. Степень прогрессирования заболевания зависит от генетического дефекта. При дистрофической миотонии у больных выявляются миотонические, миопатическиеи, эндокринные и вегетативно-трофические нарушения. В начале болезни на первый план выступают миотонические генерализованные проявления, которые постепенно угасают по мере развития парезов и атрофии, и в дальнейшем миотонический феномен проявляется в основном миотонической ямкой или валиком при перкуссии в мышцах языка. Симптомы же миопатии постепенно нарастают. Чаще всего именно миопатические проявления заставляют больных обратиться к врачу. Отмечается атрофии и слабость лицевой мускулатуры, грудино-ключично-сосцевидной мышцы и дистальных мышц конечностей. При этом выраженная атрофия височных и жевательных мышц, а также грудино-ключично-сосцевидных мышц придает характерный внешний вид больному: голова, лицо и шея, вследствие атрофии мышц, кажутся вытянутыми, истонченными в боковых направлениях. У пожилых больных часто развивается птоз и катаракта. Атрофия жевательной мускулатуры приводит к западению щек и височных ямок. Поражаются мышцы гортани и глотки, нарушается дыхания вследствие поражения межреберных мышц и диафрагмы. Атрофические поражения мышц шеи, особенно, грудино-ключично-сосцевидных мышц, вызывают на начальных стадиях запрокидывание головы назад, а при дальнейшем течении заболевания - вперед (симптом «лебединой шеи»). Вследствие поражения перонеальных мышц при ходьбе отмечаются свисание стоп. Глубокие рефлексы с конечностей угасают. Обычно при дистрофической миотонии миопатические и миотонические расстройства сочетаются с церебральными симптомами (гиперсомния, сниженный уровень интеллекта), миокардиодистрофией. С нарушением проводимости сердца и эндокринными расстройствами (нарушение менструального цикла и бесплодие у женщин; гипогонадизм и импотенция у мужчин), истончением и атрофией кожи, ранним облысением, выпадением зубов и др. По данным игольчатой ЭМГ и ЭНМГ, для дистрофической миотонии характерно сочетание миотонических высокочастотные разряды, амплитуда и частота которых волнообразно возрастают и снижаются и миопатических признаков. Активность КФК может быть несколько повышена. При ДНК-диагностике выявляется повышенное количество ЦТГ - повторов в гене DMPK. Прогноз: Клинические проявления заболевания неуклонно усиливаются, что приводит к глубокой инвалидизации пациентов. Продолжительность жизни снижена: нередко пациенты рано погибают от кардиологических осложнений, аспирационной пневмонии или дыхательной недостаточности. Миотония хондродистрофическая (син.: Болезнь Шварца–Джампеля) впервые описана Schwarz и Jampel в 1962 г. Популяционная частота неизвестна. Болеют лица обоего пола. Тип наследования аутосомно-рецессивный. Патоморфология. Может выявляться истинная гипертрофия мышц или, напротив, гипоплазия с атрофией и уменьшенными мышечными волокнами. Клиника. Больные маленького роста, имеют множественные скелетные деформации, кроме того, отмечаются миотонические проявления уже с первых лет жизни. Лицо у больных плоское, выражение печальное, рот и подбородок маленьких размеров. Аномалии глаз включают короткую и узкую глазную щель, птоз, миопию тяжелой степени, ювенальную катаракту, микрофтальмию. Характерны врожденные скелетные деформации: короткая шея, «куриная» грудь, дисплазия тазобедренного сустава, контрактуры суставов. Кроме этого, выявляют низкий рост волос, низко расположенные уши, паховые и пупочные грыжи, слабость анального сфинктера, маленькие яички, транзиторную лактозурию. Интеллект сохранен. Дифференцируют миотонию хондродистрофическую с артрогрипозом, кранио-карпотарзальной дисплазией, синдромом Секкеля, врожденной дистрофической миотонией, гипофизарным нанизмом. Холодовая парамиотония Эйленбурга (син.: врожденная парамиотония) регистрируется очень редко, передается по аутосомно-доминантному типу. Возникает вследствие мутации гена в области 17-й хромосомы. Болеют лица обоего пола. Патоморфологические изменения в мышцах такие же, как при миотонии. Патогенез до концанеизвестен, имеются указания на значительные колебания содержания калия в крови больных врожденной парамиотонией. Клиника. Болезнь может обнаружиться уже при рождении, у новорожденных по неловкости движений, например, слишком длительному закрыванию глаз при промывании их холодной водой и т. д. И в дальнейшем проявляется только под влиянием охлаждения, подчас даже умеренного, при этом развивается тоническое затруднение расслабления мышц. Сырая прохладная погода, понижение температуры в помещении до 10-12° вызывают тонические спазмы мышц. Охлаждение может быть локальным и общим. В мышцах языка (например, - после употребления мороженого и др.) может выявляться миотоническая реакция с элементами дизартрии. При охлаждении спазмы обычно поражают определенные мышечные группы - мышцы, лица, шеи, глотки, дыхательную мускулатуру, в меньшей степени конечностей. Чаще и сильнее страдают мимические мышцы; лицо пациента приобретает характерный вид: глаза прищурены, мимические мышцы находятся в состоянии тонического сокращения. Спазмы могут длиться от 15 мин. до нескольких часов. Повторные однотипные движения при врожденной парамиотонии, в отличие от миотонии Томсена, усиливают выраженность спазмов (парадоксальная миотония). Мышцы не худеют и не гипертрофируются. Под влиянием тепла спазмы обычно в течение нескольких минут исчезают. При общем охлаждении отмечаются типичные проявления миотонического феномена, а у некоторых пациентов развивается и значительная мышечная слабость, которая получила название "холодовой паралич" (врожденная парамиотония с холодовыми параличами Эйленбурга). Глубокие рефлексы с конечностей снижаются или отсутствуют. Холодовые парезы часто локализуются в проксимальных отделах конечностей, после прекращения спазмов они подчас могут длиться еще несколько дней, и в ряде случаев переходят в полный паралич. На ЭМГ в период паралича отсутствует спонтанная и произвольная активность, в периоде покоя вне охлаждения - периодические разряды спонтанной активности мышечного волокна миотонического типа, во время парамиотонического спазма - быстрые и продолжительные серии электрических разрядов. Вне охлаждения у больных выявляется только повышенная механическая возбудимость мышц. С возрастом имеется тенденция к улучшению состояния больного. В других случаяхврожденная парамиотонияможет протекать и без развития холодовых параличей (врожденная парамиотония де Йонга). Электрическая возбудимость нервов при ЭНМГ нормальная. Заболевание непрогредиентное. Диагнозмиотонииставится с учетом генетического анализа, специфических клинических феноменов, игольчатой ЭМГ, выявляющей наличие миотонических разрядов. Миотонические феномены весьма характерны, в связи с этим диагноз миотонии не представляет особой сложности. Классический тест на миотонию больного просят энергично, быстро и с усилием сжать пальцы в кулаки и быстро распрямить их. В случае миотонии сжимание происходит свободно, разжимание - медленно, с видимым напряжением, на протяжении 10-15 с. При повторных попытках миотонический феномен угасает (исключение составляет лишь парамиотония Эйленбурга, при которой, наоборот, скованность при повторных движениях усиливается). Для подтверждения диагноза проводят игольчатую ЭМГ. Дифференциальный диагноз. Миотонию дифференцируют с миотоническими синдромами при эндокринной дисфункции (микседема и др.), полимиозите, органических заболеваниях нервной системы и внутренних органов, системных заболеваниях соединительной ткани, миастении. При миотонических синдромах также выявляется и другая симптоматика, свойственная этим заболеваниям. Миотонический синдром может быть и ятрогенного генеза, в частности, вызывается приемом клофибрата. Миотонию Томсена дифференцируют с некоторыми формами миопатий (например, дюшеновской миодистрофией Беккера), а дистрофическую миотонию - со спинальной амиотрофией Верднига-Гоффмана, дистальными миопатиями, сенсомоторными полиневропатиями и др. При парамиотонии Эйленбурга дифференцируют с семейной эпизодической адинамией (болезнь Гамсторпа), нормокалиемическим периодическим параличом. При этом учитывается, что при парамиотонии Эйленбурга клинические проявления с возрастом уменьшаются. Больные приспосабливаются к дефекту и успешно работают. Трудовой прогноз при миотонии Томсена ихолодовой парамиотонии, при правильном выборе профессии в основном благоприятный, хуже - при миотонии Беккера. При дистрофической миотонии, а также примиотонии хондродистрофической прогноз неблагоприятен. Лечение миотоний.Для уменьшения выраженности миотонических проявлений назначают лекарственные препараты, стабилизирующие мембраны: фенитоин (дифенин) по 0,1-0,2 г 3 раза в день, диакарб (по 10 мг/кг/сут в течение 2-3 недель); новокаинамид, хинина, сульфат (300-400мг 3р/день), мексилетин (внутрь по 360 мг 2 раза в день), нимодипин, карбамазепин, препараты кальция. В некоторых случаях удается достичь ремиссии с помощью иммуносупрессивной терапии: в/в введение иммуноглобулина человеческого (400 мг/кг), преднизолона (1 мг/кг/сут), АКТГ, циклофосфамида. Показаны токоферола ацетат, АТФ, анаболические гормоны. Немедикаментозное лечение: электрофорез лидокаина с помощью амплипульсфореза в магнитном поле на область шейных и поясничных симпатических ганглиев, хвойные ванны, электростимуляция мышц, массаж, ЛФК. Проводят аутогенную тренировку. Рекомендуется диета с ограниченным содержанием калия. Облегчить передвижение больных можно, применяя специальные корсеты и ортезы. Прогноз. Профессиональные возможности больных ограничены, необходимо рациональное их трудоустройство. При миотонии прогноз для жизни в целом благоприятный, за исключением редких случаев дистрофической миотонии типа 1, когда возможно наступление внезапной сердечной смерти по причине кардиальной патологии. По этой причине, при дистрофической миотонии следует проводить регулярные кардиологические исследования для контроля необходимости установки постоянного электрокардиостимулятора.
ПРИЛОЖЕНИЕ Болезнь Шарко-Мари-Тута
Болезнь Шарко-Мари-Тута
Болезнь Шарко-Мари-Тута
Дата добавления: 2016-06-05 | Просмотры: 1833 | Нарушение авторских прав |