|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Резюме / Abstract
В статье описаны ЭМГ-характеристика и новые подходы в терапии наиболее часто встречающихся в практике детского невролога различных форм нейромышечных заболеваний. Представлены модифицированные стандарты диагностики и лечения детей с нейромышечной патологией, которые включают патогенетическую, специальную медикаментозную и немедикаментозную терапию (включая новые авторские технологии) с учетом степени тяжести, стадии заболевания, электромиографических, биохимических, иммунологических данных, показателей ЭКГ, холтер-ЭКГ и ЭхоКГ.
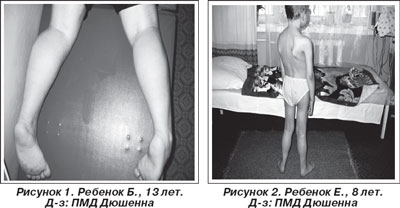
Многие заболевания неизлечимы, Нейромышечные заболевания (НМЗ) — гетерогенная в клинико-генетическом аспекте группа наследственно-дегенеративных заболеваний нервной системы, характеризующихся прогрессированием патологического процесса и приводящих к инвалидизации. В связи с увеличением числа детей с данной патологией на базе Донецкого областного детского клинического центра нейрореабилитации в 1997 году выделены амбулаторный прием и самостоятельные 4 специализированные стационарные койки для детей с нейромышечной патологией. Усовершенствована работа биохимической лаборатории, открыт ЭМГ-кабинет. Целями, связанными с реабилитацией детей с НМЗ в Центре, являются внедрение и разработка современных методов диагностики и лечения, направленного не только на замедление прогрессирования двигательных расстройств, но и на максимально возможное поддержание кардиальной и дыхательной функций. Как известно, летальный исход у большинства пациентов с различными формами НМЗ обусловлен именно вовлечением сердечной и дыхательной мускулатуры в патологический процесс, во многом определяющим характер течения заболевания и его исход. Выраженность поражения сердечной мышцы прямо пропорциональна тяжести клинической картины в статомоторной сфере. Динамическое комплексное проведение ЭМНГ, ЭКГ, холтер-ЭКГ и ЭхоКГ, спирометрии позволяет заподозрить на ранних этапах нейромышечную патологию, а также определить характер течения заболевания (медленно или быстро прогрессирующее), выявить на ранних стадиях болезни поражение сердечно-сосудистой системы, определить стадию кардиомиопатии и пневмопатии, подобрать патогенетически и симптоматически обоснованную терапию и попытаться максимально замедлить прогрессирование патологического процесса [7]. Клиническая форма заболевания устанавливается на основании комплексного (клинического, биохимического, медико-генетического, нейрофизиологического) обследования. Для диагностики ранних проявлений прогрессирующих мышечных дистрофий и амиотрофий, их фенокопий с учетом международных протоколов модифицирован диагностический и лечебный паттерн, адаптированный к практическому здравоохранению, который включает: За 12 лет (1997–2008 гг.) в базе данных Центра зарегистрировано 144 ребенка с первичной нейромышечной патологией (42 ребенка из других областей Украины, стран ближнего и дальнего зарубежья): 86 детей с миодиострофиями и миопатиями; 36 детей с наследственными полиневропатиями; 22 ребенка со спинальными амиотрофиями. Кратность прохождения курсов реабилитации в Центре, как правило, 2 раза в год, 45 % пациентов проходят курс 5–17 раз. В данной работе мы проанализировали данные ЭМГ, биохимические показатели ЭКГ и ЭхоКГ у 88 детей и попытались подобрать патогенетически и симптоматически обоснованную терапию в зависимости от степени тяжести и стадии патологического процесса. 46 детей имели миодистрофию Дюшенна. Течение: быстро прогрессирующее. Основные клинические симптомы (рис. 1, 2): слабость и атрофия мышц, преимущественно тазового, плечевого пояса, проксимальных отделов верхних и нижних конечностей; псевдогипертрофия икроножных, четырехглавых мышц, мышц предплечья; макроглоссия; когнитивные нарушения в 70 % случаев; раннее развитие контрактур, чаще в коленных, голеностопных суставах, миогенный гиперлордоз; кардио-, пневмопатии. Дебют — 2–5 лет. Тип наследования — Х-сцепленный [1, 8, 25].
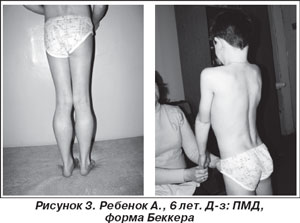
У 8 детей миодистрофия Беккера. Течение: доброкачественное. Дебют — после 5–10 лет. Основные клинические симптомы (рис. 3): нарушение интеллекта не характерно, ретракции ахилловых сухожилий, кардио-, пневмопатии выражены незначительно. Тип наследования — Х-сцепленный [25].
У 2 детей прогрессирующая миодистрофия (ПМД) Бетлема. Течение: доброкачественное, стационарное. Дебют: раннее детство. Основные клинические симптомы (рис. 4): конечностно-поясной тип распределения амиотрофий, с сохранностью лицевых мышц, контрактуры в межфаланговых, локтевых и голеностопных суставах, кардиальные проявления минимальны [1, 4].
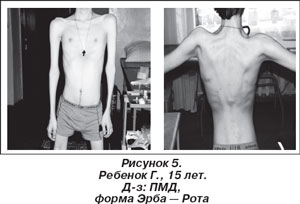
У 8 больных ПМД Эрба — Рота. Течение: злокачественное. Дебют — 3–6 лет. Основные ранние клинические признаки (рис. 5): поражение мышц тазового и/или плечевого пояса, межлопаточной области с появлением крыловидных лопаток, выраженные деформации позвоночного столба миогенного характера, преимущественно в виде поясничного гиперлордоза, кифосколиоза, прогрессирующее развитие кардио-, пневмопатий [1, 16].
У 4 детей ПМД Говерса — Веландера (дистальная миодистрофия). Течение: доброкачественное, медленно прогрессирующее. Дебют — после 4 лет. Симптоматика поражения мышц дистальных отделов конечностей. Ведущими симптомами (рис. 6) являются шлепающие стопы, слабость мышц-разгибателей кисти. Кардиальные проблемы выражены незначительно [28].
У 6 больных ПМД Эмери — Дрейфуса. Дебютирует между 4 и 15 годами жизни. Первым симптомом выступает ходьба на пальцах. Ранними и характерными признаками являются сгибательные контрактуры в локтевых суставах и разгибателях кисти, ретракции ахилловых сухожилий. Затем развиваются слабость и атрофия двуглавых и трехглавых мышц плеча, далее — в мышцах плечевого пояса. Витальный прогноз всецело зависит от степени вовлечения в патологический процесс сердечной мышцы (чаще определяется нарушение сердечной проводимости) [17]. У 5 пациентов ПМД Роттауфа — Мортье — Бейера (рис. 7). Характерной чертой болезни являются ранние, выраженные и быстропрогрессирующие сухожильные ретракции и контрактуры. От ПМД Эмери — Дрейфуса отличается более диффузным распределением мышечных дистрофий и большей скоростью прогрессирования патологического процесса. Кардиомиопатии выражены (нарушением ритма) [16, 17].
У 8 больных ПМД Давиденкова. Течение: доброкачественное. Для этой формы характерна слабость перонеальной и плечевой мускулатуры, проявляющаяся в различной последовательности или одновременно. Кардиомиопатии не типичны [23]. У 9 пациентов спинальная амиотрофия Эмери — Дрейфуса (2-й тип). Дебют: до 18 мес. Симптоматика синдрома вялого ребенка (рис. 8): генерализованная мышечная слабость, арефлексия, выраженная гипотония, быстрое и выраженное развитие костно-суставных деформаций в виде кифосколиоза, формирования вдавленной грудной клетки, контрактур в голеностопных, коленных суставах. Больные могут сидеть, в 40 % случаев — самостоятельно стоять, в 20 % случаев — ходить в ортопедической обуви без поддержки. Интеллект сохранен. Развитие кардио- и пневмопатий обусловлено грубейшей кифосколиотической деформацией позвоночного столба [1, 17].
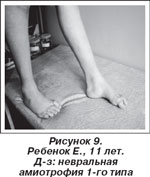
У 4 больных спинальная амиотрофия Кугельберга — Веландера (3-й тип). Дебют: после 18 мес. Течение относительно доброкачественное. Генерализованная мышечная слабость и гипотония, диффузные мышечные атрофии, преимущественно проксимальных отделов конечностей. Характерно наличие фасцикуляций в мышцах языка, пальцев, проксимальных отделов. Больные длительное время сохраняют способность стоять и ходить. Кардиальные проблемы не выражены [17]. У 1 ребенка дистальная спинальная амиотрофия. Течение медленно прогрессирующее. Заболевание начинается со слабости и атрофии передней группы мышц голени, мышц-разгибателей кисти, сочетающейся с деформацией стоп и кистей. Часто выявляется атрофия икроножных мышц [17]. У 44 детей невральная амиотрофия Шарко — Мари 1-го типа. Дебют: 1-е десятилетие жизни. Течение заболевания медленно прогрессирующее. Основные клинические симптомы: жалобы на боли в мышцах голени, чаще после физической нагрузки, затруднения при беге или подъеме по лестнице, утомляемость, частые падения. При ходьбе упор на передние отделы стоп. Восходящий тип поражения. Раньше других страдает вибрационная и тактильная чувствительность. Характерная полая стопа с молоточкообразной деформацией пальцев стопы с формированием стопы Фридрейха (рис. 9) [1, 3].
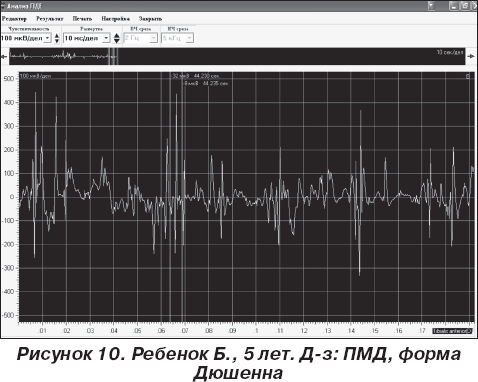
У 3 больных невральная амиотрофия Шарко — Мари 2-го типа. Клинически заболевание напоминает НМСН 1-го типа, но проявляется позднее (13–16 лет), реже вовлекаются руки, меньше выражены признаки нарушения чувствительности и деформации стопы. Одним из главных критериев, помогающим дифференцировать НМСН 2-го типа и НМСН 1-го типа, являются показатели ЭНМГ: при НМСН 2-го типа нет существенного снижения скоростей распространения возбуждения, отмечается выраженное снижение М-ответа или даже его полное отсутствие [3, 17]. Для оценки степени, преимущественной локализации, распространенности мышечной слабости, выраженности нарушений в статолокомоторной сфере, а также эффективности реабилитационного лечения у детей с НМЗ используется разработанная и адаптированная к практическому здравоохранению шкала эффективности реабилитации детей с нейромышечной патологией, включающая определение степени мышечной слабости, оценку сидения, стояния, ходьбы, подъема из положения лежа на животе, мышечного тонуса в руках, ногах. У детей со злокачественными формами прогрессирующих мышечных дистрофий отмечено снижение средней длительности потенциалов двигательных единиц (ПДЕ) на 41,2 ± 5,2 % от нормы, показатели средней амплитуды составили 358,3 ± 17,7 мкВ (при норме 600 мкВ), фазность потенциалов — 79,30 ± 5,98 % (норма до 5 %), спонтанная активность в виде потенциалов фибрилляции — 12,70 ± 0,07, положительных острых волн — 5,00 ± 0,03, псевдомиотонических разрядов — 1,30 ± 0,02 (рис. 10).
У детей с доброкачественными формами миодистрофий выявлено снижение средней длительности ПДЕ на 18,8 % ± 5,3 % от нормы, показатели средней амплитуды составили 506,7 ± 18,9 мкВ (при норме 600 мкВ), фазность потенциалов — 23,60 ± 6,12 % (норма до 5 %), спонтанная активность в виде потенциалов фибрилляции — 2,30 ± 0,08, положительных острых волн — 1,60 ± 0,02, псевдомиотонических разрядов — 0,07 ± 0,01 (рис. 11).
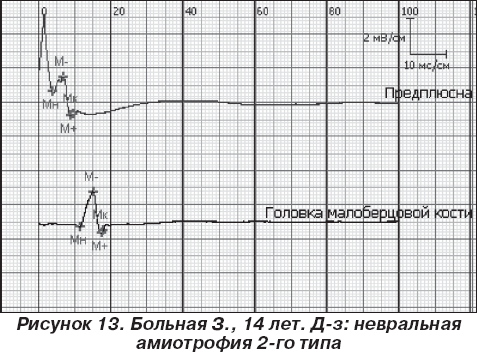
Таким образом, по данным ЭМГ с помощью концентрического игольчатого электрода удалось установить основные параметры вовлеченных в патологический процесс мышц, позволяющие определить характер и прогноз течения патологического процесса при прогрессирующих мышечных дистрофиях. Доброкачественные формы мышечных дистрофий характеризуются снижением длительности ПДЕ от 12 до 25 % нормы, снижением амплитуды ПДЕ в 1,2 раза от минимально допустимых значений, при этом фазность — до 30 %, спонтанная активность умеренно выражена в виде потенциалов фибрилляции (до 3) положительных острых волн (до 3). По данным мониторинга ЭМГ-исследований (1 раз в 6 мес.), ЭМГ-картина значительно не меняется, возможно снижение средней длительности от исходных показателей на 3–7 %, амплитуда существенно не изменяется, фазность нарастает умеренно — на 5–10 % от исходных данных, спонтанная активность часто остается без изменений. Особенно важным является выделение злокачественных форм мышечных дистрофий, которые характеризуются снижением длительности ПДЕ на 40 % и более от нормы, снижением амплитуды ПДЕ в 2 раза и более от минимально допустимых значений, фазность — от 60 до 100 %, спонтанная активность выражена в виде потенциалов фибрилляции (9 и более), положительных острых волн (6 и более), псевдомиотонических разрядов. По данным мониторинга ЭМГ-исследований (4 раза в год) отмечено дальнейшее снижение длительности ПДЕ на 20–30 % от исходных показателей, амплитуды — на 30 % и более от исходных данных, выраженное нарастание фазности потенциалов (на 30–50 % от исходного уровня), «бурная» спонтанная активность в стадии компенсации, в стадии субкомпенсации — урежение спонтанной активности на 30–45 % от предыдущих показателей и практически полное ее отсутствие в стадии декомпенсации. По электронейромиографическим и гистопатологическим показателям выделяют 2 основных типа невральных амиотрофий: миелинопатии (Шарко — Мари 1-го типа), аксонопатии (Шарко — Мари 2-го типа) [3, 9]. У детей с невральной амиотрофией Шарко — Мари 1-го типа отмечается выраженное снижение скорости проведения импульса по волокну периферического нерва вследствие прогрессирующей сегментарной демиелинизации на 30 % и более от минимально допустимого значения, увеличение длительности М-ответа, как правило, неправильной формы; моторный ответ растянут, нередко нарастает фазность, отмечается снижение терминальной латентности. По данным игольчатой ЭМГ определяются признаки стадии 3б–4 денервационно-реиннервационного процесса, которая характеризуется увеличением средней длительности ПДЕ на 12–40 % от нормы, повышением средней амплитуды ПДЕ в 1,2–2,5 раза от нормы, но увеличение фазности потенциалов происходит за счет фазности потенциалов, спонтанная активность не выявляется (рис. 12). У детей с невральной амиотрофией 2-го типа выявляются признаки аксонопатического поражения, характеризующегося снижением амплитуды на 50 % и более от минимально допустимого значения; уменьшением длительности М-ответа на 50–60 % от нормы; скорость проведения импульса по периферическим нервам долгое время может оставаться в пределах нормы; терминальная латентность, фазность М-ответа без существенных изменений (рис. 13).
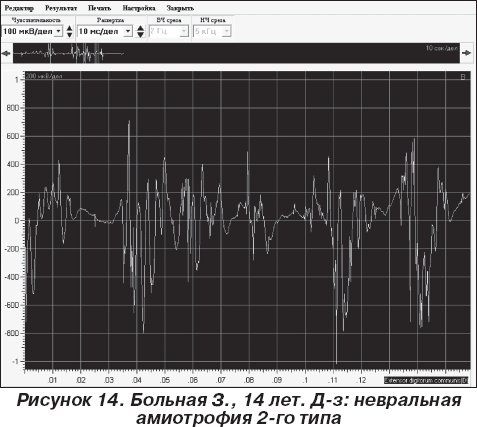
По данным игольчатой ЭМГ выявлялись признаки стадии 3б–4 денервационно-реиннервационного процесса, характеризующейся увеличением средней длительности ПДЕ на 12–40 % от нормы, повышением средней амплитуды ПДЕ в 1,2–2,5 раза от нормы, увеличением фазности потенциалов преимущественно за счет псевдополифазии; выявляется спонтанная активность в виде положительных острых волн, потенциалов фибрилляции, псевдомиотонических разрядов (рис. 14).
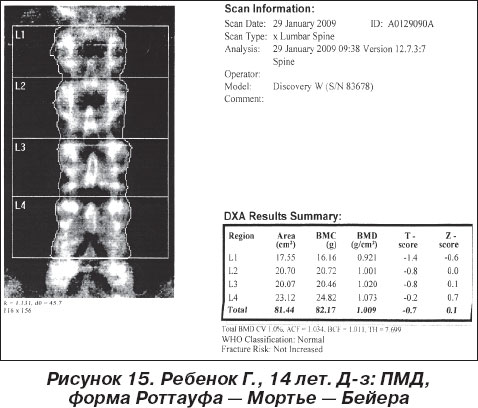
Для спинальных амиотрофий характерен нейрональный (переднероговой) тип, характеризующийся значительным увеличением длительности ПДЕ (на 40–50 % и более), амплитуды ПДЕ (до 3000–5000 мкВ при норме 350–600 мкВ); доля фазных потенциалов — от 30 % и более. По мере прогрессирования тяжести патологического процесса потенциалы становятся «гигантскими», спонтанная активность выраженная. По результатам ЭКГ- и холтер-ЭКГ-исследований, у 12 % детей выявлялась синусовая тахикардия, у 29 % — синусовая аритмия, у 35 % — укорочение интервала PQ, у 17 % — миграция источника ритма, у 14 % — суправентрикулярная экстрасистолия, у 32 % — нарушения внутрижелудочковой проводимости, у 43,5 % больных — нарушения процесса реполяризации желудочков. По данным ЭхоКГ, на стадии начальных проявлений заболевания патологические изменения выявлялись преимущественно в виде диастолической дисфункции миокарда по рестриктивному типу (уменьшение скорости потока крови на митральном клапане с уменьшением амплитуды предсердной волны, уменьшение диастолического диаметра и объема левого желудочка, умеренное снижение фракции выброса левого желудочка). На стадии субкомпенсации миопатического процесса отмечается систолическая дисфункция миокарда (умеренное увеличение систолического диаметра и объема левого желудочка, умеренное уменьшение фракции выброса). На стадии декомпенсации выявляется дилатация, гипокинезия левого желудочка (выраженное увеличение систолического диаметра и объема левого желудочка, значительное снижение фракции выброса) [13]. На основе зарубежных, отечественных и собственных данных в Центре модифицированы схемы лечения, которое направлено на полимодальное воздействие не только на костно-мышечную систему, но и на другие системы организма. Рассчитанные схемы перманентны с периодическим перерывом, обязательным повторением, сменой медикаментов и физиотерапевтических методов лечения. 1. Сбалансированное лечебное питание (продукты, содержащие белок, полиненасыщенные жирные кислоты, витамины, микроэлементы): овощи, творог, рыба, печень, соевое мясо; ограничение потребления в пищу углеводов. Как известно, ожирение является значимой клинической проблемой для пациентов с миодистрофией Дюшенна, значительно усугубляющей течение заболевания [27]. Дети хуже переносят физическую нагрузку, становятся малоподвижными, что приводит к быстро прогрессирующему формированию контрактур, атрофии сохранных мышечных волокон, развитию кардио-, пневмопатий. Поэтому, если специально подобранная диета не приносит желаемый эффект, назначали топамакс. Выбор данного антиконвульсанта обусловлен его воздействием на рецепторы Са-каналов и глутамата. По последним данным, именно выброс ионизированного Са вследствие разрушения мембран миоцитов приводит к некрозу мышечных волокон [29]. Таким образом, блокада рецепторов Са-каналов обусловливает торможение патологического процесса. Внетерапевтическим действием топамакса является подавление чувства голода. Препарат зарегистрирован Международной лигой по диете в качестве средства вспомогательной терапии при прогрессирующем ожирении. Стартовая доза составляла 1 мг/кг/сут в 2 приема, постепенно увеличивали дозу до получения клинического эффекта, но не превышая 3 мг/кг/сут, под обязательным контролем УЗИ почек и анализа мочи. Доза 3 мг/кг/сут хорошо переносится и, по нашим наблюдениям, достаточна для подавления чувства голода. Снижение веса на 30–40 % чаще происходит в течение 6 мес. По окончании приема препарата назначается строгая диета и специальная адаптированная медикаментозная терапия с ограничением назначения препаратов, способствующих нарастанию массы тела. 2. Массаж при нейромышечных заболеваниях существенно отличается от стандартных методик его проведения. Сила воздействия минимальна, акцент на улучшение трофики кожных покровов и сохранных мышц с применением актовегиновой мази, бальзама «Живокост», щадящее растягивание укороченных сухожилий с применением препаратов хондроксид, траумель С, поглаживание суставов, паравертебрально-точечный гармонизирующий массаж. Длительность сеанса — до 10 мин. Курс № 10. При наличии симптоматики слабости дыхательной мускулатуры выполняется массаж грудной клетки для облегчения дыхательных движений. Примерная схема массажа при ПМД Дюшенна: сеанс начинается с поглаживания спины, грудной клетки и конечностей с применением трофических мазей и заканчивается им, далее проводится гармонизирующий точечный массаж паравертебрально, после чего инструктор легкими движениями пальцев по ходу волокон мышц дистальных отделов конечностей прорабатывает пучки мышц, сухожилий, далее переходит на аккуратное растягивание укороченных сухожилий. Дозированная лечебная физкультура с элементами stretch-гимнастики, направленная на поддержание и максимальное сохранение функциональной способности невовлеченных в патологический процесс мышц в каждом конкретном случае с учетом формы нейромышечного заболевания. 4. Дыхательную функцию легких обследовали у 32 детей с нейромышечной патологией. 19 больных имели среднетяжелую и тяжелую степень, 13 — среднюю степень инвалидизации (модифицированная шкала оценки эффективности реабилитации детей с НМЗ). У 18 детей выявлено нарушение биомеханики дыхания по рестриктивному типу с нарушением структуры общей емкости легких. У 24 детей отмечалось снижение показателей жизненной емкости легких: легкое — у 16, умеренное — у 4, значительное — у 3, весьма значительное — у 1. Повышение показателей остаточного объема легких выявлено у 28 больных: незначительное — у 12, умеренное — у 11, значительное — у 4, резкое — у 1. В комплекс лечения пневмопатий включена синглетно-кислородная терапия (valkion-терапия). Физико-химическая концепция valkion-терапии базируется на фотохимической сенсибилизации воздуха и воды с образованием вторичных долгоживущих физиологически активных форм кислорода и оксида азота — valkion-факторов, способствуюших активации клеточного метаболизма, уменьшению гипоксии тканей, восстановлению слизистой бронхов, нормализации функции внешнего дыхания, улучшению дренажной функции бронхов [7]. Использовали следующую схему: 1-й день — 100 мл воды, 5 мин ингаляции, 2–3-й день — 150 мл воды, 9 мин ингаляции, 4-й и последующие дни — 200 мл воды, 14 мин ингаляции. За 30 мин до процедуры дети принимали перорально поливитамины с содержанием витаминов А, Е и С. Курс № 10–15, 3–4 курса в год. Дети и родители обучаются специально разработанной в Центре дыхательной гимнастике, вокалотерапии, в основе которой лежит произношение звуков во время активного выдоха с акцентом на гласные звуки. В домашних условиях рекомендуется надувание резиновых шаров. Применение бронхолитиков-адреномиметиков (сальбутамол, вентолин и др.) у больных с крайне ограниченной двигательной функцией приводит к расслаблению гладкой мышцы, резкому повышению уровня креатинкиназы, миоглобина, что может вызвать острую почечную недостаточность. А гипокалиемия вызывает нарушение работы сердца. Поэтому применяемая доза должная использоваться с осторожностью. 5. Сопутствующая патология со стороны опорно-двигательного аппарата: деформация туловища, позвоночника (нейромышечный сколиоз) — увеличивает тяжесть дыхательных расстройств, в связи с чем в Центре применяется механизированная кровать без подогрева, воздействие которой имитирует легкую мануальную терапию. Для укрепления мышечного корсета спины паравертебрально применяется вакотрон — импульсная электротерапия током низкой частоты с использованием вакуумного электрода, что значительно снижает время воздействия процедуры и усиливает ее эффект. Вакуумное наложение электродов позволяет существенно ускорить сам процесс наложения электродов. Включение импульсного режима разряжения усиливает кровообращение, и, как результат, увеличивается эффективность процедур электротерапии. К вакотрону может быть подключено 4 вакуумных электрода диаметром 60 или 90 мм. Степень разряжения регулируется бесступенчато от минимального показателя до 0,6 бар. Установленное значение поддерживается автоматически. Время воздействия 5–10 мин. Курс № 10. 6. Для профилактики контрактур применяются специальные шины, валики, метод фиксации конечностей в физиологическом положении на ночь с использованием туторов. С целью адаптации передвижения с оптимальной коррекцией деформаций используются стельки, ортопедическая обувь, наколенники, на протяжении 1–2 часов носят реклинаторы, корсеты в моменты наибольшей нагрузки на позвоночный столб (сидение, ходьба и др.). В стадии декомпенсации корсеты необходимо носить практически постоянно, поскольку клинически имеет место выраженная атония и гипотрофия мышечного корсета, влекущие за собой резкую деформацию позвоночного столба, что может привести к вторичной висцеропатии, ухудшению работы сердца, легких, что, в свою очередь, обусловливает еще большую декомпенсацию патологического процесса [10, 32]. При доброкачественных формах нейромышечных заболеваний (ПМД Бетлема, Эмери — Дрейфуса, Роттауфа — Мортье — Бейера) в стадии компенсации возможно проведение оперативных вмешательств, направленных на предупреждение контрактур, сухожильных ретракций и избавление от них, коррекцию деформаций [7]. С целью укрепления мышечного корсета спины использовали фармакопунктуру паравертебрально с применением микродоз (0,1 мл на точку). При спинальных, невральных амиотрофиях применяли 0,5% раствор нейромидина. При миодистрофии Дюшенна, сочетающейся с когнитивными нарушениями, применяли фармакопунктуры с кортексином (церебролизином). Количество инъекций 10. 7. Наличие остеопороза, выявленного у большинства больных с миопатиями и амиотрофиями [5], обусловливает назначение кальцийсодержащих препаратов (кальций-D3) на протяжении периода до 6 мес., затем 1 мес. перерыв, в этот промежуток рекомендуется диета, обогащенная солями кальция (нежирный творог, сыворотка, измельченная яичная скорлупа, твердые сорта сыра), с обязательным контролем ионизированного Са крови, проведением денситометрии костей (рис. 15, наши исследования).
8. В связи с частыми ОРВИ необходимо назначение препаратов, влияющих непосредственно на иммунокомпентные клетки и центральные механизмы регуляции иммунитета, через которые оказывается вторичное иммуностимулирующее влияние на организм. К таким препаратам относятся: бронхо-мунал П, рибомунил, иммунал, ИРС. Бронхо-мунал П назначался на протяжении 10 дней с 20-дневным перерывом детям до 12 лет по 3,5 мг/сут, детям старше 12 лет — по 7,0 мг/сут. Курс № 3. 9. У детей с прогрессирующими мышечными дистрофиями и амиотрофиями мышечное волокно неспособно поддерживать мышечное сокращение заданной интенсивности, поэтому необходимы препараты, действие которых направлено на энергетическое обеспечение сохранной мышечной ткани и функциональная активность которых сопряжена с энергетическим пулом ацетил-КоА в митохондриях. Препараты метаболического действия назначаются в сочетании или отдельно, курсами до 3 месяцев 2 раз в год: В лечении спинальных амиотрофий целесообразно назначение препаратов нейротрофического действия, улучшающих метаболизм и микроциркуляцию спинного мозга: Каждый год появляются новые данные о лечении нейромышечных заболеваний. Несмотря на генетическую теорию, в меньшей степени касающуюся этиологии, в большей — патогенеза, в частности, в 2003 году появились данные о влиянии вальпроевой кислоты на РНК мутантного белка у детей с аутосомно-рецессивной формой спинальной мышечной атрофии [19, 31]. Как известно, вследствие мутации происходит исключение 7-го экзона псевдогена SMN2 из продукта транскрипции, что приводит к продукции неполноценного рибосомального белка [3]. По данным мировой литературы [19, 31], вальпроевая кислота воздействует именно на ядерные белки и рибонуклеопротеиды 7-го экзона. Это в конечном итоге приводит к увеличению концентрации нейротрофического белка [20], что оказывает положительное влияние на течение спинальной амиотрофии 2-го типа у детей. Больные получали вальпрoаты в дозе 15–30 мг/кг массы тела [19, 20, 31]. Положительная динамика в виде нарастания силы, увеличения переносимости физических нагрузок, снижения амплитуды, длительности, фазности ПДЕ, спонтанной активности, по данным ЭМГ, отмечалась после 7–9-го мес. приема препарата. Стойкий эффект сохранялся в среднем 3–4 мес. после окончания приема препарата [19, 20, 31]. Безусловно, определенные нововведения, по-видимому, ограничатся меньшими дозами из-за мышечно-релаксирующего эффекта антиконвульсантов [11]. У 2 из 3 детей с СМА 2-го типа применялся конвулекс 10 мг/кг. За 12 месяцев наблюдения отмечена стабилизация неврологического дефицита. В определении тактики лечения невральных амиотрофий любого типа помогают ЭМГ-данные, позволяющие определить преимущественный тип поражения нервного волокна (аксональный, демиелинизирующий, смешанный). При миелинопатиях показано назначение одного из нижеописанных препаратов: При аксонопатии назначали: Возможно назначение препаратов с разными механизмами действия (например, нейромультивит + нуклео ЦМФ форте; нейромидин + нуклео ЦМФ форте; кортексин + нейромультивит) и обязательно в сочетании с препаратами метаболического действия (кудесан, элькар, стимол и др.). При дилатационных кардиомиопатиях показано курсовое внутривенное введение кардиотрофических препаратов: Вместе с тем проблема интенсивного прогрессирования патологического процесса при нейромышечных заболеваниях остается актуальной. Одной из попыток решения данной проблемы является гипотеза «механизма контроля, регуляции синтеза и секреции интерлейкинов-1, -6, -4, -10 [12], а также определенной взаимосвязи с тепловым обменом организма, т.е. с температурой тела ребенка». Наблюдая за 144 детьми в течение 10 лет, мы обратили внимание, что у 22 больных (ПМД — 13 человек, СМА — 9 человек) во время ОРВИ, протекавшей с высокой температурой тела (38–38,5 °С) в течение 3–5 дней и у 9 (из 22) человек после прошедших катаральных явлений через 3—5 дней отмечено улучшение мышечной активности и общего самочувствия. Мы попытались объяснить данный феномен. Наша гипотеза была подкреплена тем, что генетическая структура рибосомального гена идентифицируется как один из локусов гена SMA, наиболее часто подверженного повреждению при амиотрофии [3, 20], для «запуска» образования рибосомального белка/цитокина требуется определенный стимул [5] (в частности, повышение температуры тела). Согласовав вопрос с этическим комитетом и получив разрешение родителей для временного повышения температуры у детей, мы применили обезболивающий препарат пирогенал — липополисахарид, образующийся в процессе жизнедеятельности микроорганизмов. Область применения препарата пирогенал — стимулирование восстановительных процессов после травм спинного мозга; посттравматические невропатии; рассасывание патологических рубцов, спаек после ожогов; в комплексном лечении больных инфекционными заболеваниями с затяжным, рецидивирующим течением; бронхиальная астма, псориаз, нейродермит [5, 22]. Пирогенал применен у 4 детей (3 девочки, 1 мальчик) в возрасте от 5 до 8 лет. Из них ПМД Беккера страдало 2 человека, СМА 2-го типа — 2 человека. Дозы подбирались индивидуально. Вводили препарат внутримышечно 1 раз в сутки через день. Начальная доза составляла 5–15 минимальных пирогенных доз (МПД), устанавливали дозу, вызывающую повышение температуры тела до 37,5–38 °С, и вводили до прекращения повышения температуры тела, после чего дозу постепенно увеличивали на 10–25 МПД, но не более 250–500 МПД. Курс лечения — 15 инъекций. Перерыв между курсами — 2–3 мес. Положительная динамика выражалась в виде увеличения переносимости физических нагрузок, нарастания силы. Положительный потенциал зафиксирован от 3 до 5 мес., на фоне перорального приема метаболических препаратов: кудесана, элькара, стимола. Катамнез наблюдения — 1 год. Ухудшения состояния в контроле через 12–15 мес. по основному заболеванию ни у одного ребенка не отмечено. Рассмотрим аутоиммунный процесс с позиции возможной коррекции не только стимулирования защитных свойств организма, но и с точки зрения провоспалительных механизмов за счет повышения уровня IgG [7]. Мы попытались соединить достижения нейроиммунологии и миологии [15]. Как известно, Деструкция мышечных волокон — перманентный процесс, запускаемый определенными генами и характерный для злокачественно текущих ПМД. На определенном этапе заболеваний возникает аутоиммунный процесс, больше связанный с тонкими механизмами иммунопатологии, в частности с продукцией интерлейкинов. Интерлейкины-1, -6, ФНО-α обладают провоспалительным эффектом, т.е. способствуют активации аутоиммунного процесса, в то же время интерлейкины-4, -10 обладают противовоспалительным действием [12, 22]. Именно наличие повышенного уровня провоспалительных интерлейкинов диктовало необходимость проведения коррекции у наших больных. Прием препарата биовен моно, в котором высока концентрация IgG, способствует увеличению количества собственых IgG и депрессии вторично развивающейся аутоиммунной реакции. Таким образом, внутривенный иммуноглобулин активизирует противовоспалительные цитокины и косвенно активизирует трофические функции по данным ФНО-α. Основные заболевания нервной системы, при которых эффект применения иммуноглобулина доказан: атаксия-телеангиэктазия (синдром Луи-Бара); синдром Гийена — Барре; хронические демиелинизирующие воспалительные полиневропатии. Заболевания нервной системы, при которых применение иммуноглобулина эффективно: энтеровирусный менингоэнцефалит; тяжелая миастения; мультифокальные невропатии; антифосфолипидный синдром (преходящие нарушения мозгового кровообращения, инсульты, васкулопатии); рассеянный склероз; рефрактерные формы эпилепсии у детей. Заболевания нервной системы, при которых применение иммуноглобулина замедляет прогрессирование заболевания: дерматомиозит; миодистрофия Дюшенна; диабетическая полиневропатия [29]. Основными механизмами иммуномодулирующего действия иммуноглобулина для внутривенного введения являются: нейтрализация патогенных аутоантител антиидиопатическими антителами, подавление провоспалительных цитокинов, ингибирование комплемента и предотвращение образования мембранолитического комплекса, уменьшение числа естественных клеток-киллеров и подавление экспрессии антигена-1 на поверхности Т-лимфоцитов [22, 24]. По данным иммунограммы, у детей с миодистрофиями отмечается снижение абсолютного и относительного количества Т3-лимфоцитов — 72 %; показателей Т8-супрессоров — 59 %; IgG — 94 %; CD20 — 37 %; СD95 (фактор апоптоза), повышение относительного количества Т4-хелперов — 63%, достоверно увеличение интерлейкина-4 (до лечения 7,48 ± 0,37, после лечения 11,61 ± 0,55, р < 0,01, при норме 14,9 ± 1,2), интерлейкина-10 (до лечения 2,17 ± 0,05, после лечения 3,25 ± 0,05, р < 0,05, при норме 3,73 ± 0,1). Иммуноглобулин человека нормальный применялся нами в дозе 5 мл/кг на инфузию. Весь объем иммуноглобулина разводится в 3–4 раза изотоническим раствором и вводится со скоростью 20–25 капель в минуту. Количество инфузий — от 3 до 5, одновременно назначаются малые дозы преднизолона по 5–10 мг утром 1 р/д сроком на 3–6 мес. Курсы внутривенного введения иммуноглобулинов повторяют 4 раза в год (каждые 3 мес.). Одновременно перорально дети принимали преднизолон. Преднизолон — катаболический стероид, механизм действия которого основан на противовоспалительном и иммуносупрессивном воздействии при длительном приеме, в то же время короткие курсы обладают стимулирующим эффектом. Некоторые исследователи предполагают [21], что преднизолон стимулирует образование дистрофиноподобных белков, кроме того, замедляет ослабление легочной функции, увеличивает фракционной выброс крови левым желудочком. Как известно, применение малых доз преднизолона (0,5–1 мг/кг) оказывает стимулирующий, а не супрессирующий эффект, в то время как при демиелинизирующих заболеваниях (рассеянный склероз, энцефалиты и др.) назначается пульс-терапия — 10 мг/кг. Результаты внутривенного введения иммуноглобулина на фоне перорального приема преднизолона: из 10 обследованных детей с генетически подтвержденной мышечной дистрофией Дюшенна положительная динамика отмечена у 4 детей (40 %). При анализе биохимической картины крови (КФК, ЛДГ, АЛТ, АСТ) наблюдается снижение показателей, отражающее стабилизацию клеточных мембран миоцитов. По данным иммунограммы отмечена нормализация абсолютного и относительного числа Т-лимфоцитов, повышение показателей Т-супрессоров, тенденция к снижению и нормализации относительного количества Т-хелперов, показатели СD95 увеличились (по нашим данным, этот показатель может служить определенным маркером для лечения иммуноглобулином детей с миодистрофиями). Катамнез — 2 года. Ухудшения состояния по основному заболеванию ни у одного ребенка не отмечено. Достигнутый положительный потенциал поддерживается путем приема элькара, кудесана, нейромидина. Таким образом, при более достоверной диагностике с учетом тонкостей электромиографических вариантов течения патологического процесса сегодня возможно говорить о замедлении прогрессирования нейромышечной патологии при таких формах, как ПМД Дюшенна, Эрба — Рота, спинальная амиотрофия 2-го типа. Это является достаточно существенным достижением с точки зрения улучшения качества жизни. Поэтому разработка схем лечения больных с начальными проявлениями и на стадии прогрессирования, новых технологий, в частности применения иммуноглобулинотерапии, кортикостероидов, метаболических и антиоксидантных средств, требует дальнейшего накопления материала исходя из опыта положительного влияния на лечения. В настоящее время диагностика ПМД и СМА существенно опережает их терапию. Поэтому патогенетический метод лечения данных заболеваний не должен сводиться только к физио-, кинезотерапии и наблюдению, оказанию помощи по показаниям. Не вылечить — не значит помочь. Перенимая опыт западно-европейских коллег в терапии ПМД и СМА, все же назрела необходимость аналитического осмысления достижений и отечественных исследований. Интеграция отечественных генетиков существенно продвинет решение данной проблемы.
Дата добавления: 2014-12-11 | Просмотры: 1371 | Нарушение авторских прав |






 Основное содержание статьи изложено в лекции, прочитанной на Международной научно-практической конференции, посвященной современным стандартам диагностики и лечения детей с нейромышечной патологией (21.05.2009 — 23.05.2009, Харьков, Украина). Лекция вызвала интерес и внимание слушателей. Во время дискуссии отечественными исследователями был затронут вопрос о целесообразности применения преднизолона при миодистрофии Дюшенна, назначении антиоксидантов, метаболических и других средств, возможности применения в/в иммуноглобулина при прогрессирующих формах миодистрофий и пирогенала при спинальных амиотрофиях 2-го типа. Применение преднизолона при миодистрофии Дюшенна было подтверждено иностранными гостями. В частности, Томас Сьорсен (Швеция) подробно остановился на дозах преднизолона при миодистрофии Дюшенна, сославшись на положительный результат исследований Европейской сети нейромышечной патологии Treat-NMD. В перерыве конференции проф. Волкера Штрауба (Великобритания) и Томаса Сьорсена (Швеция) заинтересовала методика применения valkion-терапии при миодистрофиях и пирогенала при спинально-мышечной атрофии 2-го типа (поскольку в странах Европы существует сертифицированный препарат пироген). Согласно международному опыту, в Швеции некоторым больным проводится курсовое внутривенное введение иммуноглобулина, однако он крайне дорог. В данной работе при помощи глубоких клинико-инструментальных, биохимических и иммунологических (в ряде случаев — генетических, а не ex juvantibus) исследований проводился подбор медикаментозного лечения. В личной беседе с иностранными учеными (практиками) был выделен интересный факт: антиоксиданты, метаболическая терапия, нейропротекторы за рубежом применяются в виде специальных диет или небезызвестного канадского коктейля, содержащего сбалансированный набор витаминов, антиоксидантов и метаболических средств. В нашей стране это в определенной степени компенсируется назначением медикаментозных и немедикаментозных препаратов, содержащих антиоксиданты, нейротрофические и метаболические средства. Но, к глубокому сожалению, без генетической идентификации невозможно продвижение вперед и участие в международных исследованиях и апробации новых лекарственных средств.
Основное содержание статьи изложено в лекции, прочитанной на Международной научно-практической конференции, посвященной современным стандартам диагностики и лечения детей с нейромышечной патологией (21.05.2009 — 23.05.2009, Харьков, Украина). Лекция вызвала интерес и внимание слушателей. Во время дискуссии отечественными исследователями был затронут вопрос о целесообразности применения преднизолона при миодистрофии Дюшенна, назначении антиоксидантов, метаболических и других средств, возможности применения в/в иммуноглобулина при прогрессирующих формах миодистрофий и пирогенала при спинальных амиотрофиях 2-го типа. Применение преднизолона при миодистрофии Дюшенна было подтверждено иностранными гостями. В частности, Томас Сьорсен (Швеция) подробно остановился на дозах преднизолона при миодистрофии Дюшенна, сославшись на положительный результат исследований Европейской сети нейромышечной патологии Treat-NMD. В перерыве конференции проф. Волкера Штрауба (Великобритания) и Томаса Сьорсена (Швеция) заинтересовала методика применения valkion-терапии при миодистрофиях и пирогенала при спинально-мышечной атрофии 2-го типа (поскольку в странах Европы существует сертифицированный препарат пироген). Согласно международному опыту, в Швеции некоторым больным проводится курсовое внутривенное введение иммуноглобулина, однако он крайне дорог. В данной работе при помощи глубоких клинико-инструментальных, биохимических и иммунологических (в ряде случаев — генетических, а не ex juvantibus) исследований проводился подбор медикаментозного лечения. В личной беседе с иностранными учеными (практиками) был выделен интересный факт: антиоксиданты, метаболическая терапия, нейропротекторы за рубежом применяются в виде специальных диет или небезызвестного канадского коктейля, содержащего сбалансированный набор витаминов, антиоксидантов и метаболических средств. В нашей стране это в определенной степени компенсируется назначением медикаментозных и немедикаментозных препаратов, содержащих антиоксиданты, нейротрофические и метаболические средства. Но, к глубокому сожалению, без генетической идентификации невозможно продвижение вперед и участие в международных исследованиях и апробации новых лекарственных средств.