|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
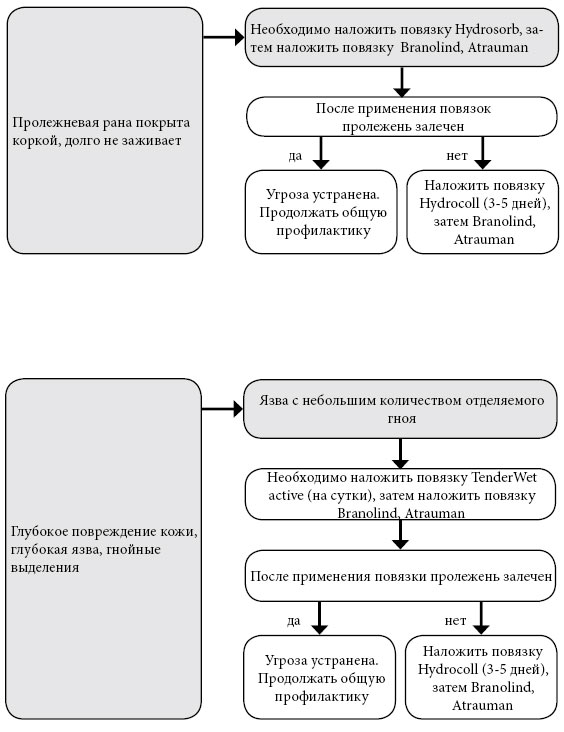
Фенилкетонурия. Повязка Hydrocoll (самоклеящаяся) наклады
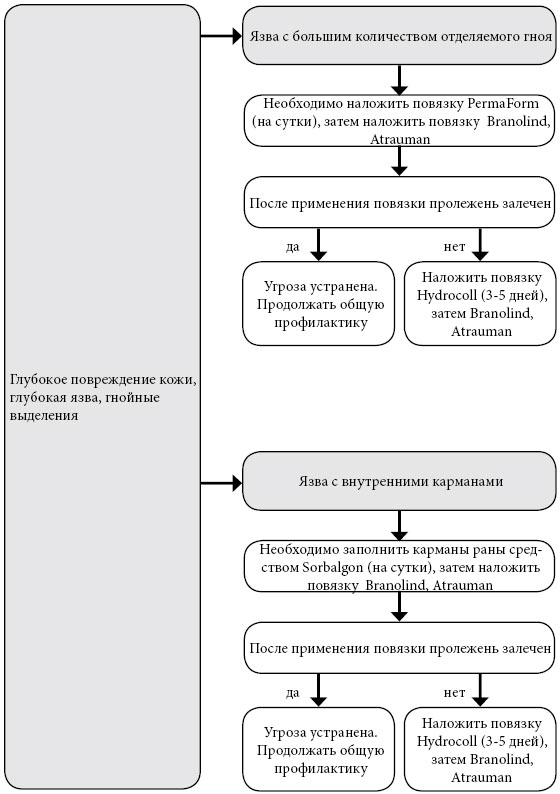
Повязка Hydrocoll (самоклеящаяся) накладывается на 3-8 дней, впитывает (абсорбирует) экссудат и превращает его в гель, гель поддерживает в ране влажную среду. Способствует грануляции и эпителизации поврежденного участка. Сигналом к смене повязки служит ее деформация в форме пузыря. Повязка Atrauman оказывает бактерицидное действие и ускоряет заживление раны. Представляет собой сетку, покрытую тончайшим слоем серебра и пропитанную гидрофильной мазью. Повязка впитывает экссудат, уничтожает бактерии в ране и способствует регенерации кожи.
Повязка TenderWet activity в форме мягкой подушечки, пропитанной раствором Рингера, обеспечивает непрерывное «промывание» раны в течении 24 часов. В результате применения повязки некротические ткани отторгаются, происходит регенерация сосудов и образование грануляционной ткани. Повязка Hydrosorb создает в ране оптимальную влажную среду, впитывает излишний экссудат, способствует отторжению некротической ткани, ускоряет заживление. Повязка Sorbalgon является высокоэффективным средством очищения и заживления пролежней с гнойным отделяемым. Повязка представляет собой ватообразный материал, который вводится в карман обширной пролежневой язвы. Если в кармане гнойное дно, Sorbalgon впитывает его в себя, очищает рану, затвердевает и удаляется из кармана легким нажатием на область вокруг раны. Если дно кармана чистое, повязка превращается в гель, очищает рану и самостоятельно вытекает из нее. Повязку можно использовать для тампонирования глубоких ран. Повязка PermaForm из инновационной губчатой матрицы втягивает содержимое раны и не выпускает обратно (экссудат, гной). Одновременно повязка создает благоприятную для заживления среду и стимулирует образование грануляционной ткани. Удаляется повязка безболезненно и без остатков. Размер и способы крепления повязок варьируются. К каждой повязке прилагается инструкция, в которой прописано, должна ли повязка выходить за края раны, как ее фиксировать и как долго можно не снимать. Самоклеящиеся повязки по мере необходимости безболезненно удаляются. Мазевые повязки и повязки без крепления фиксируются специальным пластырем или бинтом. Специальные пластыри на бумажной основе (Hartmann Omnifilm, Cosmopor E, Omnifix Elastic) предпочтительнее, так как они не перекрывают дыхание кожи и при снятии не отрывают клетки верхнего слоя эпидермиса. Фиксируя повязку, старайтесь сильно не натягивать пластырь, чтобы не образовались нежелательные складки кожи. Если пролежень глубокий, то его необходимо на всю глубину рыхло (но не туго) тампонировать (закрыть) повязкой. За кожей вокруг пролежня нужно тщательно ухаживать, применяя средства госпитальной гигиены. Во время гигиенических процедур кожу нельзя тереть, а только слегка промакивать, обязательно высушивая полотенцем.
Фенилкетонурия ЧТО ТАКОЕ ФЕНИЛКЕТОНУРИЯ? Фенилкетонурия, которую сокращенно называют ФКУ, является одной из наследственных болезней обмена веществ, обусловленной изменением (мутацией) в определенном гене. ФКУ не очень редкое заболевание. В России оно встречается в среднем с частотой 1 на 7000 новорожденных. КАК НАСЛЕДУЕТСЯ ФЕНИЛКЕТОНУРИЯ?
У бабки по матери мутантный ген также имеется только в одной хромосоме, как и у деда со стороны отца. Они, как и родители больного ребенка, здоровы, но передали хромосомы, содержащие мутантный ген, своим детям. У вторых деда и бабки обе хромосомы содержат только нормальный ген. Таким образом, при рецессивном наследовании болен только тот член семьи, который получил от своих родителей обе хромосомы, несущие мутантный ген. Все остальные члены семьи здоровы, в том числе и те, кто является носителем мутантного гена. На представленном фрагменте родословной видно, что у родителей больного ребенка могут еще появиться больные дети. Вероятность появления больного ребенка в семьях, в которых родители являются носителями мутантного гена, составляет 1/4 или 25%. Эта вероятность не меняется от числа больных или здоровых детей в семье: для каждого следующего ребенка риск, что он будет болен, составляет 25%. Вероятность рождения здорового ребенка, обе хромосомы которого содержат только нормальный ген, составляет также 25%. Вероятность появления детей, у которых будет один нормальный и один дефектный ген, составляет 50%, они, как и их родители, будут здоровыми носителями мутантного гена. Многие родители больных ФКУ детей и их родственники, первый раз встретившись с врачом-генетиком, настойчиво повторяют, что у их ребенка не наследственное заболевание, так как в их семье ни у кого из родственников никогда не было такого заболевания. В этом случае только доступное объяснение о том, что правила наследования бывают разные, и не редко больной с наследственным заболеванием бывает единственным в семье, позволяют родителям понять, с какой ситуацией они столкнулись. ПОЧЕМУ РЕБЕНОК МОЖЕТ ЗАБОЛЕТЬ ФЕНИЛКЕТОНУРИЕЙ? ФКУ обусловлена мутацией в гене, который отвечает за синтез фермента, участвующего в превращении аминокислоты фенилаланина в тирозин. В результате мутации в гене фермент оказывается дефектным, фенилаланин не может превратиться в тирозин и накапливается в крови. Возникает так называемый метаболический блок. Фенилаланин, как и тирозин, являются аминокислотами, а аминокислоты - это те кирпичики, из которых построены все белки. Так как новорожденный постоянно получает с пищей белки (в молоке матери основным белком является казеин), то уровень фенилаланина постоянно растет и, наконец, достигает таких концентраций, при которых он становится токсичным, в первую очередь, для развивающегося мозга младенца. КАКИЕ НАРУШЕНИЯ В ОРГАНИЗМE ВЫЗЫВАЕТ ФЕНИЛКЕТОНУРИЯ? Без лечения у 95% младенцев с ФКУ развивается тяжелая умственная отсталость и задержка в моторном развитии. Дети не могут сидеть, стоять, ходить, и умственное развитие с возрастом продолжает снижаться. Кроме того, у больного могут появиться судороги, экзема на коже, а в старшем возрасте присоединяются грубые нарушения в поведении. У некоторых больных отмечается маленький размер головы и сердечные пороки. ЧТО ТАКОЕ СКРИНИНГ НОВОРОЖДЕННЫХ НА ФЕНИЛКЕТОНУРИЮ? Для того, чтобы избежать развития таких тяжелых клинических проявлений ФКУ нужно, чтобы новорожденный с повышенным содержанием фенилаланина в крови был выявлен в первые дни жизни. Для этого практически во всех странах мира существуют программы скрининга на ФКУ. В России скрининг новорожденных проводится в абсолютном большинстве территорий. Он заключается в том, что у новорожденного на 4 -5 день жизни перед выпиской из родильного дома берут из пятки несколько капель крови, которую наносят на специальную фильтровальную бумагу. Кровь высушивается, и такой бланк, на котором указана фамилия новорожденного и ряд других сведений, необходимых для его идентификации, переправляется в лабораторию региональной медико-генетической консультации. В лаборатории определяют содержание в крови уровня фенилаланина. Если уровень фенилаланина оказывается низким, то это означает, что у ребенка нет ФКУ. Если же уровень фенилаланина в крови высокий, то возникает подозрение на фенилкетонурию. В этом случае лаборатория запрашивает теперь уже в педиатрической службе, так как новорожденный выписан из родильного дома, повторное взятие крови у младенца. В этой связи родители узнают от педиатра, что первый тест на ФКУ у их ребенка оказался ненормальным. У них появляется повод для беспокойства. Повторное тестирование образца крови у ребенка является решающим. В большинстве случаев при повторном исследовании уровень фенилаланина оказывается нормальным. Это означает, что результат первого исследования был неверный (его называют ложноположительным). Причины этого могут быть связаны как с состоянием младенца на момент взятия крови, так и с какой-то ошибкой в анализе. Этот результат, свидетельствующий о том, что у ребенка нет ФКУ, сразу сообщается родителям, и они могут успокоиться. ЧТО ДЕЛАТЬ, ЕСЛИ ДИАГНОЗ ФЕНИЛКЕТОНУРИИ ПОДТВЕРДИЛСЯ? Если и при втором тестировании уровень фенилаланина остается высоким, то это означает, что ребенок болей ФКУ, и семья немедленно приглашается в медико-генетическую консультацию. Здесь родителям объясняют, что такое ФКУ, почему она возникла у их ребенка, и назначают лечение. Если лечение начато рано, то клинические симптомы ФКУ у ребенка не проявятся, и он будет расти здоровым, практически не отличаясь от сверстников. Смысл лечения заключается в уменьшении содержания фенилаланина в пище, которую получает ребенок. Обычно это достигается за счет специальных смесей, содержащих в необходимых количествах все незаменимые аминокислоты, за исключением фенилаланина. Успех лечения во многом определяется тем, насколько родители больного ребенка осознали важность диетотерапии, и насколько строго они ее выполняют. Обо всем этом и о многом другом семье расскажет врач- генетик во время первого визита семьи в медико-генетическую консультацию. Затем такие визиты станут регулярными. У ребенка будут постоянно контролировать содержание фенилаланина в крови, и, в зависимости от лабораторных показателей, корректировать состав тех продуктов, которые, с одной стороны, не будут повышать уровень фенилаланина, а с другой, обеспечивать нормальный рост и развитие ребенка. Постоянный контакт семьи с врачом-генетиком является залогом успешного лечения фенилкетонурии. В России диетотерапия для больных ФКУ проводится до возраста ребенка 7 - 8 лет, но многие родители продолжают ее и дольше. МОЖНО ЛИ ПОМОЧЬ СЕМЬЕ, В КОТОРОЙ ПОЯВИЛСЯ БОЛЬНОЙ ФКУ, ИМЕТЬ ЗДОРОВЫХ ДЕТЕЙ? Да, и довольно успешно. Для ФКУ возможна дородовая диагностика. Первым шагом в этом направлении является обращение в медико-генетическую консультацию, где врач-генетик определяет показания и возможные методические подходы к дородовой диагностике. В каждом конкретном случае решается вопрос о необходимости молекулярно-генетического обследования больного ребенка или родителей, а затем плода. Сама процедура заключается в том, что во время беременности в сроке 9 - 11 недель или 16 - 18 недель врач акушер-гинеколог проводит забор очень небольшого количества клеток плода, находящихся в околоплодной жидкости, плодных оболочках или крови плода, и направляет этот материал в специальную лабораторию пренатальной диагностики. В этой лаборатории врачи лаборанты-генетики проводят молекулярную диагностику, т.е. определяют наличие или отсутствие мутации в гене, отвечающем за фенилкетонурию. В случае положительного результата семья решает вопрос о прерывании беременности больным плодом или настраивается на появление еще одного больного ребенка. Это право выбора остается за семьей. Дата добавления: 2015-01-18 | Просмотры: 1150 | Нарушение авторских прав |

 Повязка Branolind (мазевая, сетчатая), пропитанная перуанским бальзамом, обладает антисептическим и ранозаживляющим действием.
Повязка Branolind (мазевая, сетчатая), пропитанная перуанским бальзамом, обладает антисептическим и ранозаживляющим действием. Наследуется ФКУ по аутосомно-рецессивному типу, т.е. больные накапливаются в семье в одном поколении. Схема такого наследования приведена на рисунке, на котором изображен фрагмент родословной семьи, в которой родился ребенок, больной ФКУ. На родословной мужчины обозначены квадратиком, а женщины - кружочком. Внутри этих квадратиков и кружочков нарисована только одна хромосома из 23 пар, имеющихся у человека. Эта хромосома несет нормальный или дефектный (мутантный) ген фенилкетонурии, последний помечен черной точкой.
Наследуется ФКУ по аутосомно-рецессивному типу, т.е. больные накапливаются в семье в одном поколении. Схема такого наследования приведена на рисунке, на котором изображен фрагмент родословной семьи, в которой родился ребенок, больной ФКУ. На родословной мужчины обозначены квадратиком, а женщины - кружочком. Внутри этих квадратиков и кружочков нарисована только одна хромосома из 23 пар, имеющихся у человека. Эта хромосома несет нормальный или дефектный (мутантный) ген фенилкетонурии, последний помечен черной точкой.