|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Аномалии в системе половых хромосом
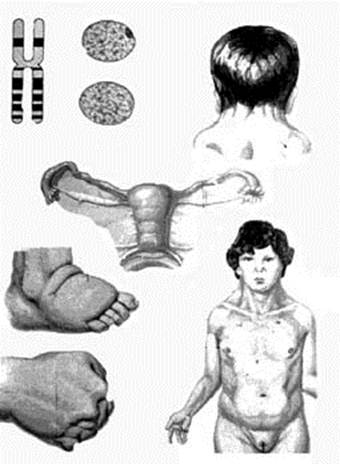
Синдром шерешевского-тернера (моносомия Х; 45, XО)
Для синдрома характерно отсутствие в кариотипе половой Х-хромосомы (рис.10). Частота встречаемости 1:3000, среди девочек, страдающих олигофренией – 1:1500. Частота синдрома возрастает среди низкорослых женщин с недоразвитием вторичных половых признаков и аменореей. Большинство больных с синдромом Шерешевского–Тернера имеют нормальный или близкий к норме интеллект, но умственная отсталость у них встречается чаще, чем в общей популяции. Интеллектуальные нарушения обычно сочетаются с недоразвитием эмоционально-волевой сферы: больные повышенно внушаемы, несколько некритичны, упрямы, часто эйфоричны. Диагностика синдрома возможна уже в период новорожденности: · Девочки рождаются с низкой массой тела и небольшого роста; · Отмечается отечность кистей и стоп; · Низкий рост волос на шее, шея короткая с крыловидными складками, идущими от сосцевидных отростков к плечам; · Характерна чрезмерная подвижность кожи на шее.
Отмечаются множественные аномалии развития: · Эпикант, антимонголоидный разрез глаз; · Низко расположенные ушные раковины; · Гипомимия («лицо сфинкса»); · Микроретрогнатия, высокое небо, аномалии зубов. · Важными диагностическими признаками являются также врожденные пороки сердца, низкий рост (в 98% случаев), половой инфантилизм с первичной аминореей, часты гипоплазия или гипертрофия ногтевых пластинок, гиперпигментация кожи. Наблюдаются дефекты зрения (22%) и слуха (52%) Характерны разнообразные скелетные нарушения · «Щитообразная» широкая грудная клетка; · Гипоплазия или сращение I и II шейных позвонков; · Широкие кисти с короткими IV и V пальцами; · Деформация локтевых и коленных суставов; · Укороченные III и IV пальцы стоп, синдактилия. Офтальмологическое обследование выявляет бледность сосков зрительного нерва, микрофтальм, катаракту, сужение артерий глазного дна. Дерматоглифическое исследование выявляет изменение кожных узоров пальцев и ладоней. Диагноз может быть установлен с помощью цитологического метода исследования полового хроматина и кариологического анализа.
Синдром трисомии (47, XXX)
Для синдрома характерно наличие в кариотипе дополнительных Х-хромосом. Частота трисомии Х среди новорожденных девочек 1:800. Частота возрастает среди пациенток психиатрических больниц. В период новорожденности и детства редко можно выявить какие-либо фенотипические особенности, имеющие диагностическое значение. Основная психопатологическая особенность синдрома – проявление эмоциональной незрелости и эмоционально-поведенческие нарушения с невротическими и неврозоподобными расстройствами, иногда со склонностью к аутоагрессии. В раннем возрасте характерно выраженное отставание в развитии речи. У женщин с трисомией Х часто наблюдается эндокринный дисбаланс, бесплодие, преждевременный климакс. Могут наблюдаться более сложные полисомии Х: тетрасомия (ХХХХ) и пентасомия (ХХХХХ). Считается, что степень психического недоразвития коррелирует с числом дополнительных Х-хромосом. У женщин с полисомией Х увеличена частота психических заболеваний (шизофрения, эпилепсия, маниакально-депрессивный психоз). Окончательный диагноз устанавливается на основании цитологического обследования щечного эпителия в результате обнаружения полового хроматина в кариотипе.
Синдром клайнфельтера (47, XXY)
Для синдрома характерно наличие в кариотипе мужчины дополнительной половой Х-хромосомы. Частота синдрома составляет в среднем 1 на 850 новорожденных мужского пола и 1–2.5% у больных олигофренией в степени дебильности. Клинические проявления достаточно вариабельны. Обязательными диагностическими критериями являются гипогенитализм и гипогонадизм. Характерными признаками также являются: · Высокий рост, высокое стояние таза; · Евнухоидные пропорции, астеническое телосложение, узкие плечи, удлиненные конечности. Мышечная система развита слабо. Это особенно четко проявляется в препубертантном и пубертантном возрасте. У взрослых нередко встречаются склонность к ожирению по женскому типу, гинекомастия, слабое подмышечное оволосение, оволосение на лобке по женскому типу. Отмечают недоразвитие вторичных половых признаков с гипоплазией яичек и часто полового члена. При гистологическом исследовании яичек выявляют гиалиноз и фиброз семенных канальцев. При исследовании спермы обнаруживаются олиго- или азооспермия, в пунктате яичка – гиперплазия клеток Лейдига, гиалинизация семенных канатиков, кроме того, характерен высокий уровень фолликулостимулирующего гормона. Мужчины с синдромом Клайнфельтера бесплодны. Частыми являются различные диспластические признаки: · Брахицефалия; · Низкий рост волос на затылке, уплощенный затылок; · Гипертелоризм; · Эпикант; · Деформация ушных раковин; · Выступающие надбровные дуги; · Аномалии зубов; · Искривление и укорочение V пальцев. Болезнь часто сопровождается задержкой психического развития. Диагноз может быть установлен на основании кариологического анализа, обнаружения полового хроматина в щечном эпителии.
Синдром дубль Y (47, XYY)
Синдром характеризуется наличием в кариотипе дополнительной Y-хромосомы. Наблюдается у мальчиков и мужчин высокого роста. Частота среди новорожденных мальчиков 1:840. Выраженных нарушений фенотипа может не наблюдаться. Примерно у 80% лиц с данным синдромом наблюдаются признаки психического недоразвития в сочетании с нарушениями эмоционально-волевой сферы и поведения. Больные испытывают трудности в социальной адаптации. Многим характерны замедленность и ригидность мышления, речи и моторики, часто снижена способность к самокритике. Наблюдается сочетание умственной отсталости с психопатоподобным поведением, агрессивностью, расторможеностью и извращением влечения. Отмечаются самоуверенность, импульсивность, гиперсексуальность. Окончательный диагноз устанавливается при цитологическом обследовании. Высказывается предположение, что психопатоподобные формы поведения при наличии несбалансированного кариотипа по половым хромосомам связаны с вторичными изменениями в деятельности нервной системы как следствие нарушений гормональной сферы. Признанными причинами хромосомных нарушений считаются ионизирующая радиация, тяжелые инфекции и интоксикации, эндокринные нарушения, воздействие химических веществ и общее загрязнение окружающей среды. Большая роль в профилактике хромосомных синдромов принадлежит медико-генетическому консультированию. Окончательный диагноз хромосомного заболевания ставится на основании цитогенетических исследований кариотипа. Ориентировочный диагноз может быть поставлен с помощью экспресс-метода (определение полового хроматина в эпителии слизистой оболочки щеки). Среди диагностических тестов антропометрических методов значительное место занимает дерматоглифика (от греч. derma – кожа glipho – гравирую). Кожные узоры на пальцах и ладонях закладываются с третьего месяца внутриутробной жизни. К концу четвертого месяца их формирование завершается полностью, и в течение всей дальнейшей жизни узоры остаются постоянными. Определение некоторых синдромов с помощью метода дерматоглифики имеет высокую степень достоверности. 1. Трисомия - 13 (синдром Патау) – при анализе ладонных узоров наблюдаются дистальные осевые трирадиусы (угол – atd=180’), радиальные петли на четвертом и пятом пальцах рук, увеличение удельного веса дуг в узорах пальцев рук и ног, окончание главной линии А у радиального края ладони, наличие дополнительных узоров на гипотенаре, дуговые или Т-образные узоры на поле большого пальца. 2. Трисомия - 18 (синдром Эдвардса) – дуги не менее чем на шести пальцах, а в 80% случаев – на всех пальцах рук и ног. На пятом пальце, а иногда и на всех пальцах отсутствует дистальная сгибательная складка, на поле большого пальца стопы – дуговой узор. 3. Трисомия - 21 (синдром Дауна) – дистальное смещение осевого трирадиуса (L atd=81’), ульнарная петля на втором пальце и радиальная на четвертом и пятом пальцах, увеличение удельного веса ульнарных петель на пальцах, отсутствие дистальной сгибательной складки на мизинце, учащение узоров на гипотенаре, снижение общего гребневого счета TRC, непрерывность папиллярных линий. 4. Синдром «кошачьего крика» (46.5р–) – увеличение удельного веса завитков и дуг, уменьшение радиальных петель, наличие промежуточного осевого трирадиуса, горизонтальное направление папиллярных линий, уменьшение общего числа гребневого счета TRC. 5. Синдром Клайнфельтера – наиболее часты увеличения удельного веса дуг, снижение гребневого счета, проксимальное смещение осевого трирадиуса, повышение частоты узора на гипотенаре. 6. Синдром Шерешевского - Тернера – отмечается увеличение удельного веса завитков и уменьшение удельного веса дуг, учащение узора на гипотенаре, вертикальная направленность линий ладоней, снижение частоты узоров на тенаре.
ВОПРОСЫ И ПРАКТИЧЕСКИЕ ЗАДАНИЯ
1. Какие гаметы могут образовываться у человека, если патология нерасхождения по 21 хромосоме наблюдалась: (1) в анафазе I, (2) в анафазе II мейоза? 2. Какое число хромосом будет в гаметах человека, если нерасхождение по двум негомологичным хромосомам произошло: (1) в анафазе I, (2) в анафазе II мейоза? 3. В потомстве каких организмов следует ожидать большего генетического разнообразия: размножающихся вегетативным или половым путем? Почему? 4. Нормальное число хромосом в клетках человека равно 46. Сколько хромосом содержат сперматозоиды, яйцеклетки, полярные тельца? 5. Составить схему гаметогенеза у мужской особи, гомозиготной по гену А (или а), начиная со стадии сперматогонии, имеющего одну пару хромосом. 6. Составить схему гаметогенеза у гетерозиготной мужской особи Ав, начиная со стадии сперматогонии, имеющего одну пару хромосом. 7. Составить схему гаметогенеза у мужской особи, гомозиготной по двум парам генов А (или а) и В (или в), начиная со стадии сперматогонии, имеющего две пары хромосом. Негомологичные хромосомы изобразить различными по форме и величине. 8. Составить схему гаметогенеза мужской дигетерозиготной особи АаВв, начиная со стадии сперматогонии, имеющего две пары хромосом. Негомологичные хромосомы изобразить различными по форме и величине. 9. Сколько аллелей одного гена может содержаться в зрелой половой клетке? 10. Определить количество полового хроматина в соматических клетках женщины, содержащей три половые хромосомы – ХХХ. 11. Определить количество полового хроматина в соматических клетках мужчины, содержащих три половые хромосомы – ХХY. 12. Ребенок с синдромом Дауна имеет 46 хромосом вместо 47, обычно обнаруживаемых при этом заболевании. Исследование его кариотипа показало, что одна из его хромосом (№ 15) длиннее обычной. У матери больного, а также у бабушки по материнской линии (с нормальной конституцией) обнаружены 45 хромосом с удлиненной хромосомой № 15. Чем можно объяснить наблюдаемое в этой семье явление? 13. Мать имеет 45 хромосом, так как одна из 21 пары хромосом транслоцирована на 15 (т.е. 15/21), отец имеет нормальный кариотип. Какие по генотипу могут образоваться зиготы у этих родителей и какова их дальнейшая судьба? 14. Определить название мутаций в приведенных кариотипах, указать, в каких хромосомах они произошли: a) 46, XX, 1 pter 22; b) 46, XY, X q28; c) 46, XY, t (13; 21); d) 46, XY, 8 qinv 12/22. 15. Какие из следующих заболеваний не связаны с нарушением мейотического нерасхождения хромосом: · Синдром Тернера; · Синдром Дауна; · Синдром «кошачьего крика»; · Синдром Патау. НАСЛЕДСТВЕННЫЕ ГЕННЫЕ БОЛЕЗНИ
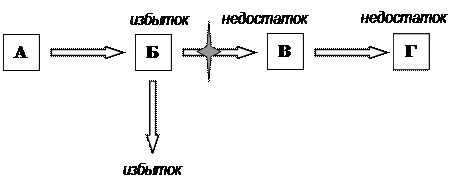
Наследственные генные болезни обусловлены генными мутациями, изменяющими генетический код синтеза белков. Генные мутации возникают, когда последовательность нуклеотидов в ДНК гена изменяется. Существуют два основных класса генных мутаций: замена пар нуклеотидов, когда одна или несколько нуклеотидных пар в ДНК заменяются другими; мутация со сдвигом рамки считывания, обусловленные вставкой или выпадением одного или нескольких нуклеотидов. Замены пар оснований в нуклеотидной последовательности структурного гена часто приводят к замене одной аминокислоты в полипептидной цепи, определяемой одним геном. Мутации со сдвигом рамки считывания сильно изменяют последовательность аминокислот в транслируемом белке. Нарушение синтеза белка при мутации соответствующего гена приводит к количественному или качественному изменению белка в организме. Генные мутации у человека являются причинами многих форм наследственной патологии. Если изменяется белок–фермент, выполняющий каталитическую функцию, то нарушается сложная цепь превращения вещества в организме: ген → фермент → биохимическая реакция → признак. В биологической литературе такого рода изменения принято называть биохимическими мутациями, в медицинской литературе их называют наследственными дефектами обмена веществ или наследственными энзимопатиями. Функциональная неполноценность ферментной системы ведет к резкому нарушению определенного биохимического процесса или биохимическому блоку. Метаболический блок можно определить по накоплению в организме вещества, которое образуется на стадии, предшествующей этому блоку (схема 1). Выпадение одного единственного метаболического звена приводит к серьезным вторичным расстройствам обмена веществ и к множественным патологическим изменениям в организме.
Степень снижения активности фермента может быть разной как при различных энзимопатиях, так и при данной энзимопатии. Снижение активности фермента или его отсутствие может быть обусловлено разными мутациями, происходящими в разных кодонах гена. Кроме того, снижение активности фермента может быть связано с мутационным дефектом одного из компонентов ферментной системы. Следовательно, одни и те же биохимические изменения могут быть вызваны аллельными мутациями или мутациями в нескольких неаллельных генах. Таким образом, одна и та же энзимопатия может иметь несколько генетических форм. Это явление получило название генетической гетерогенности. Широкая генетическая гетерогенность энзимопатии в значительной мере определяет изменчивость их клинических проявлений. Однако только особенностями мутационного гена нельзя объяснить неодинаковое проявление болезни у разных больных. В значительной степени ген проявляется во взаимосвязи с другими генами, вне зависимости от передающихся в семье. Эти гены могут усилить или затормозить проявление основного гена. Они могут изменить феномен наследственной болезни. Основной ген, в свою очередь, влияет на проявление других генов, благодаря чему у больного могут выявляться дополнительные, несвойственные основному заболеванию симптомы. Таким образом, эффект мутантного гена можно рассматривать, как многоступенчатый процесс, первой ступенью которого является первичный биохимический дефект, второй – вовлечение в процесс других ферментных систем и развитие сложных метаболических расстройств, третий – формирование клинического феномена болезни. Следует, однако, помнить, что проявление действия гена в целостном организме в определенной степени зависит также от индивидуального состояния ряда функциональных систем и факторов внешней среды, которые могут применять проявление отдельных симптомов болезни. Особенно опасно, если такие факторы оказывают свое действие в самом раннем возрасте, когда мозг ребенка особенно чувствителен к различным биохимическим нарушениям. По количеству затронутых мутацией генов выделяют моногенные и полигенные болезни. Моногенные болезни обусловлены мутацией в одном гене. Полигенные болезни обусловлены сложным взаимодействием многих генов с факторами среды. Для наследственных моногенных болезней характерны три типа наследования: аутосомно–рецессивный, аутосомно–доминантный и Х–сцеплен-ный.
Моногенные болезни, наследуемые по аутосомно–рецессивному типу
Для аутосомно–рецессивного типа наследования характерно: · Мутантный ген проявляется только у гомозигот по рецессивному гену. · Если родители гетерозиготны, то вероятность рождения больного ребенка составляет 25%. · При анализе родословной мутантный ген проявляется не в каждом поколении. · Вероятность проявления мутантного гена возрастает в родственных браках. · Частота проявления мутантного гена у лиц женского и мужского пола одинакова.
Фенилкетонурия (ФКУ) – наследственное заболевание обмена, характеризующееся поражением ЦНС и прогрессирующим, особенно в первые 2–3 года жизни, слабоумием. Фенотипически здоровые родители больного ребенка являются гетерозиготными носителями мутантного гена. Частота заболевания в Европе в среднем составляет 1: 10 000 новорожденных, распространенность носителей гена в популяции 1: 50. ФКУ наблюдается примерно у 1% умственно отсталых лиц: чем тяжелее в социальном плане контингент обследуемых, тем чаще выявляют заболевание. Заболевание обусловлено мутацией гена, контролирующего синтез фермента фенилаланингидроксилазы, который обеспечивает превращение поступающего в организм с пищей фенилаланина в тирозин (схема 2). Нарушение последнего процесса приводит к резкому повышению содержания фенилаланина в сыворотке крови и спинномозговой жидкости, при этом отмечают дефицит тирозина, что определяет недостаточный синтез катехоламинов, гормона щитовидной железы и меланина, при недостаточном количестве которого наблюдается слабая пигментация кожи и волос. При ФКУ нарушается также обмен триптофана и синтез серотонина, что губительно действует на нормальное функционирование нервной системы. Ген РАН локализован на хромосоме 12q22. Дети с ФКУ рождаются с полноценным головным мозгом, так как биохимические процессы плода осуществляются за счет процессов в организме матери. Возникающие после рождения биохимические нарушения оказывают токсическое воздействие на нервную систему, в результате чего нарушаются процессы миелинизации, развитие и рост мозга. Нарастание интеллектуального дефекта сочетается с отставанием в физическом развитии, часто с признаками умеренной микроцефалии. Характерен внешний вид больных (блондины со светлой кожей и голубыми глазами) и отдельные диспластические признаки (высокое небо, эпикант, деформация ушных раковин). При этом отмечают следующие неврологические нарушения: мышечную гипертонию, повышение сухожильных рефлексов, гиперкинезы, тремор пальцев рук, атаксию, нарушения черепно–мозговой иннервации. В более редких случаях имеет место мышечная гипотония; судорожный синдром наблюдается у 20–50% больных.
Уровень интеллектуального развития колеблется от нормы до глубокой идиотии. Прогредиентность динамики слабоумия наиболее выражена в первые 2–3 года жизни. Больные отличаются инертностью, недостаточной целенаправленностью с характерными нарушениями внимания, памяти, недоразвитием гностических функций и пространственных представлений. Отмечается также выраженное недоразвитие речи и нарушения звукопроизношения. Нарушения речи обычно сопоставимы с глубиной интеллектуального дефекта. Гомоцистинурия обусловлена отсутствием или снижением активности фермента цистатионинсинтетазы, необходимого для синтеза цистатионина из гомоцистеина и серина. Этот метаболический блок представлен на схеме 3. Ген локализован на хромосоме 21q22.
Дефект генетически гетерогенен. Существуют две формы, различающиеся по отношению к витамину В6: пиридоксинзависимая и пиридоксинрезистентная. Описаны случаи заболевания, вызванные дефицитом других ферментов. Частота заболевания среди новорожденных колеблется от 1:80 000 до 1:180 000. Среди умственно-отсталых частота гомоцистинурии достигает 0.3%. В контингенте умственно отсталых с дефектами зрения – 2.6%. Клиническая картина полиморфна, но вместе с тем наиболее типичным симптомокомплексом считается сочетание умственной отсталости с дефектами зрения (эктопия хрусталика, катаракта, миопия) и костной системы (удлинение трубчатых костей при укороченном туловище, деформация суставов, вальгусная деформация стоп, крыловидные лопатки). Внешними, наиболее выраженными признаками являются мягкие светлые волосы, голубые радужки, диспропорциональность телосложения с укорочением туловища и удлинением конечностей в сочетании со многими стигмами дизэмбриогенеза (воронкообразная грудная клетка, остеопороз костей и др.). Поражение соединительной ткани, механизм которого еще не ясен, определяет сходство гомоцистинурии с болезнью Марфана. Существует предположение о патогенетической роли в патогенезе гомоцистинурии дефицита меди. Нервно–психические нарушения при этом заболевании отмечаются в 75% случаев. Описаны легкие (пограничные) и глубокие формы умственной отсталости с инертностью нервных процессов, недостаточной критичностью, расстройством речи. В ряде случаев отмечены двигательные нарушения в виде параличей и парезов. Нарушения речи включают общее недоразвитие, косноязычие, дизартрию. Биохимическая диагностика направлена на качественное определение цистина и гомоцистина в моче, а также количественное определение метионина и гомоцистина в плазме на аминокислотном анализаторе. С целью ПД определяется активность цистатионинсинтетазы в культуре амниотических клеток. Лечение заключается в диете, бедной метионином. При пиридоксинзависимой форме заболевания эффективна терапия большими дозами витамина В6. Галактоземия обусловлена нарушением обмена галактозы. Этот метаболический путь представлен на схеме 4. Ген, контролирующий синтез фермента галактозо-1-фосфат-уридилтрансфераза локализован на хромосоме 9р13. Генная мутация в гене приводит к дефициту фермента и к биохимическому блоку на этапе галактозо-1-фосфат. Биохимический катагенез болезни включает накопление галактозы и галактозо–1–фосфата в разных тканях и в крови. Вторичным эффектом является нарушение использования глюкозы в печени, почках и головном мозге.
В выраженных случаях клинические проявления отмечаются уже с первых дней жизни ребенка в виде расстройств пищеварения и признаков интоксикации (гипотрофия, рвота, понос, отказ от кормления), желтухи с увеличением размеров печени, двусторонней врожденной катаракты. Иногда катаракта возникает несколько позже – на 4–7-й неделе жизни. При некоторых моносимптомных формах эти проявления выражены нерезко, отмечаются либо умственная отсталость, либо катаракта в сочетании с непереносимостью молока. В более тяжелых случаях наблюдается сложный дефект – сочетание умственной отсталости с нарушением зрения (слепота). При рано начатом лечении диетой дети могут развиваться нормально. Диагностируют галактоземию с использованием комплекса диагностических средств (в настоящее время создана система ее раннего выявления). Для предупреждения тяжелых нервно–психических отклонений разработана безлактозная диета. Синдром Ушера. Распространенность синдрома Ушера среди детей с глубокой глухотой составляет от 3 до 10%. По данным европейского семинара по синдрому Ушера (1997 г.) люди с этим заболевание составляют до 6% всех глухих с рождения и до 50% всех слепоглухих взрослых. Потеря зрения выявляется обычно в возрасте около 10 лет. Нарушение зрения медленно прогрессирует. Полная слепота может наступить в 50–60 лет. Офтальмологическое обследование обнаруживает типичный медленно прогрессирующий пигментный ретинит. Пигментный ретинит начинается скоплением гранул пигмента на глазном дне, распространяющихся по направлению к периферии. Поля зрения медленно сужаются и параллельно снижается острота зрения. К другим глазным симптомам относятся катаракта, глаукома. Выявляется врожденная нейросенсорная потеря слуха от умеренной до резко выраженной степени. У больных отмечается атрофия кортиева органа и эпителия внутреннего и наружного желобка в нижней части базального завитка улитки. Дегенеративные изменения в верхнем завитке. Имеется резкая атрофия спирального узла, его периферических и центральных волокон. Обнаруживаются дефекты вестибулярной системы, которые выражаются в нарушении равновесия при ходьбе. Нарушение равновесия возможно обусловлено нарушениями лабиринта, а не мозжечковой патологией. У больных, кроме основных симптомов, выявляются также психозы, агрессивность, периодические депрессии, у 25% - умственная отсталость. Синдром наследуется по аутосомно-рецессивному типу. Ген локализован на хромосоме 14q. Сочетание глухоты с пигментным ретинитом впервые было описано А. Графе в 1858 г., а генетическую природу этого синдрома установил С. Ушер в 1914 г. Выявлено, что один из 100 человек является носителем гена синдрома Ушера. У гетерозигот могут быть выявлены отсутствие реакции на вращение, повышение порога темновой адаптации или незначительное снижение зрения. Своевременное выявление у больных пигментого ретинита и создание адекватных педагогических условий предотвращают стрессовые состояния, связанные у глухого человека с потерей зрения. Методы лечения отсутствуют. Дата добавления: 2015-01-12 | Просмотры: 3081 | Нарушение авторских прав |