|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
ФВ: флаконы с порошком по 0,5 и 1,0Кетамин (Ketamine, Calypsol). Кетамин можно рассматривать как производное мощного галлюциногенного наркотического средства фенциклидина. Механизм действия: Кетамин связывается с фенциклидиновым участком аллостерического центра NMDA-рецептора и блокирует его. NMDA-рецепторы сопряжены с кальциевыми каналами на мембране нейрона, их эндогенным лигандом является глутаминовая кислота, которая обеспечивает открытие канала. При блокаде аллостерического центра рецептора его сродство к глутаминовой кислоте резко падает и обычные концентрации медиатора уже неспособны активировать рецептор и открыть канал. Прекращение тока ионов кальция через пресинаптическую мембрану нарушает экзоцитоз медиатора, а прекращение тока ионов кальция через постсинаптическую мембрану – нарушает генерацию длительных возбуждающих потенциалов. Основное влияние кетамина осуществляется на рецепторы таламокортикальных областей мозга, которые принадлежат ретикулярной фармации. В итоге, устраняется стимулирующее влияние этой системы на корковые структуры. ФК: после внутривенного введения наркоз возникает уже через 1-2 минуты и продолжается 15-20 мин. После внутримышечного введения скорость наступления наркоза замедляется (возникает через 3-4 мин), но его длительность увеличивается до 40 мин. Кратковременность действия кетамина обусловлена его перераспределением из ткани мозга в жировую и мышечную ткань, а также деметилированием в печени. ФЭ: 1. Кетамин вызывает диссоциативную анестезию, которая характеризуется сочетанием анальгезии (потерей болевой чувствительности), амнезии (потерей памяти на период действия кетамина) и кататонии (обездвиженностью) при сохранении сознания у пациента. 2. Кетамин не снижает мышечный тонус и не вызывает миорелаксации, напротив, он способен несколько усиливать рефлексы (в том силе гортанные и глоточные), что затрудняет проведение интубации трахеи. 3. После применения кетамина возникает длительная анальгезия (около 3-4 часов), полагают, что это связано с образованием в печени фармакологически активного метаболита норкетамина. В большей степени кетамин понижает соматическую и в меньшей степени висцеральную чувствительность, поэтому его редко применяют при операциях на внутренних органах. 4. Кетамин – единственный анестетик, который способен оказывать стимулирующее влияние на силу сердечных сокращений, увеличивать АД и СВ. Полагают, что это связано с его способностью нарушать обратный захват катехоламинов. В итоге повышается уровень катехоламинов в синапсах симпатической нервной системы и усиливается ее влияние на миокард и артериальные сосуды. Поэтому кетамин часто применяют у пациентов с гипотонией, после кровопотери. Чаще всего кетамин применяют в качестве вводного наркоза, изредка – для поддержания наркоза при относительно непродолжительных операциях. НЭ: 1. Кетамин усиливает мозговой кровоток, повышает внутричерепное давление и потребность мозга в кислороде. 2. Повышение артериального давления на фоне кетамина может привести гипертоническому кризу. 3. Кетамин усиливает выход ионов калия из мышц, поэтому его нельзя сочетать с деполяризующими миорелаксантами (сукцинилхолином), которые также увеличивают выделение калия из мышц. При совместном применении кетамина и сукцинилхолина возможно развитие гиперкалиемии и остановка сердца. 4. Галлюцинаторный синдром – возникает после выхода из наркоза, характеризуется дезориентацией в пространстве, красочными зрительными галлюцинациями, которые носят иногда устрашающий характер, осязательными галлюцинациями. Данный эффект может быть предупрежден введением дроперидола или диазепама. ФВ: ампулы 5% раствора по 2 и 10 мл. Натрия оксибутират (Natrii oxybutyras, GOBA). Является натриевой солью g-оксимасляной кислоты. Механизм действия: ГОМК легко проникает через ГЭБ в ЦНС, где превращается путем транаминирования в эндогенный медиатор g-аминомасляную кислоту. ГАМК активирует ГАМКА-рецепторы, которые сопряжены с хлоридными каналами. Поступление ионов хлора в клетку вызывает гиперполяризацию постсинаптической мембраны (снижается возбудимость нейрона). Сама по себе ГОМК является агонистом ГАМКВ-рецепторов, которые локализованы на пресинаптической мембране холинергических и адренергических синапсов и тормозят выделение медиатора в синаптическую щель. ГАМК-рецепторы располагаются главным образом в корковых структурах мозга, поэтому ГОМК влияет преимущественно на кортикальные функции и рефлексы спинного мозга. ФК: после внутривенного введения наркоз возникает через 0,5 ч и продолжается до 1,5-3,0 часов. При внутримышечном или пероральном введении скорость наступления наркоза замедляется (развивается через 1 час), но его длительность существенно не меняется. ФЭ: 1. Введение ГОМК вызывает наркоз с выраженной миорелаксацией, но неполным выключением рефлексов, поэтому достаочно часто ГОМК применяют при хирургическом осмотре у маленьких детей, это позволяет устранить сопротивление ребенка врачебному осмотру, но при этом сохраняются диагностически значимые рефлексы. 2. В малых дозах ГОМК применяют иногда в расчете на успокаивающее и снотворное действие. 3. ГОМК оказывает антигипоксическое действие (повышает устойчивость ткани к гипоксии). Полагают, что это связано с тем, что часть ГОМК превращается в янтарный полуальдегид и состема ГОМК/полуальдегид выступает в роли альтернативного переносчика протонов в обход дыхательной цепи митохондрий, устраняя ацидоз и накопление недоокисленных продуктов. 4. ГОМК практически не влияет на работу дыхательного, сосудодвигательного центра, уровень АД и ВЧД. 5. ГОМК оказывает слабое ноотропное действие (улучшает когнитивно-мнестические функции нервной системы). В настоящее время ГОМК применяется достаточно редко. Обычно это средство используют для поддержания наркоза, при обезболивании родов, у лиц с черепно-мозговыми травмами. НЭ: ГОМК относительно малотоксичное средство. При быстром внутривенном введении возможно подергивание мышц конечностей и языка, развитие рвоты. Во время выхода из наркоза иногда наблюдается двигательное и речевое возбуждение. ФВ: ампулы 20% раствора по 10 мл ПРОТИВОПАРКИНСОНИЧЕСКИЕ СРЕДСТВА Жена же Лотова оглянулась позади его, и стала соляным столпом Бытие, 19:26 Противопаркинсоническими называют лекарственные средства, которые применяют для лечения болезни или синдрома Паркинсона. Паркинсонизм (болезнь Паркинсона, дрожательный паралич) – хроническое прогрессирующее заболевание при котором поражаются ядра экстрапирамидной нервной системы. Клиническая картина Паркинсонизма включает 4 основных синдрома: · Ригидность мышц – повышение тонуса скелетных мышц и затруднения при совершении пассивных движений; · Олигокинезия – заторможенность, обеднение движений (возможна олигомимия - маскообразное лицо, лишенное мимики); брадифрения – психическая и аффективная (чувственная) заторможенность; · Тремор – стереотипное ритмичное дрожание головы и рук, которое усиливается в покое и исчезает во время сна; · Синдром вегетативных нарушений – слюнотечение, повышенная потливость, сальность кожи (эти симптомы обусловлены преобладанием тонуса блуждающего нерва). Развитие болезни Паркинсона сопровождается изменением психики человека – возникает психическая заторможенность, депрессия, мышление пациента вязкое, речь монотонная с преобладанием уменьшительно-ласкательных оборотов, возможны быстрые переходы от благодушия к дисфории. Физиология и патофизиология экстрапирамидной системы. Экстрапирамидная система представлена 3 основными центрами: paleostriatum (бледным шаром), neostriatum (хвостатое ядро и скорлупа) и s. nigra (черное вещество).
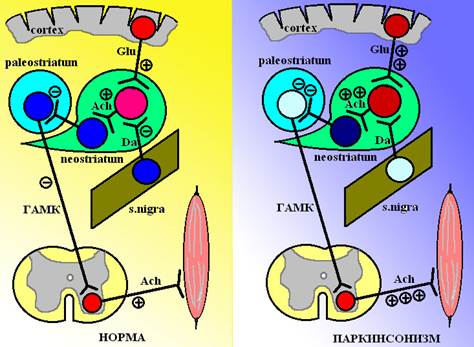
Схема 1. Слева представлены нормальные взаимоотношения между компонентами экстрапирамидной системы: наблюдается балланс дофамин- и холинергических влияний на нейроны хвостатого ядра (neostriatum). Справа представлена картина, которая имеет место у пациента с паркинсонизмом: усиление холинергических влияний на фоне недостатка тормозных дофаминергических импульсов. Ach – ацетилхолин, Glu – глютаминовая кислота, Da – дофамин. В норме холинергические a-мотонейроны спинного мозга находятся в состоянии постоянной активности и повышают тонус мышц. Функция мотонейронов спинного мозга тормозится ГАМК-ергическими нейронами бледного шара через ГАМКВ-рецепторы, расположенные на a-мотонейронах (при этом тонус мышц понижается). В свою очередь, хвостатое ядро также имеет ГАМК-ергические нейроны, которые тормозят бледный шар, при этом прекращается угнетение мотонейронов спинного мозга и тонус мышц повышается. Полноценный контроль мышечного тонуса в экстрапирамидной системе осуществляется путем взаимодействия возбуждающих глутаматергических нейронов коры, тормозных дофаминергических нейронов черного вещества и возбуждающих холинергических нейронов хвостатого ядра. Во время физической активности глутаматергические нейроны коры головного мозга через NMDA-рецепторы стимулируют холинергические нейроны хвостатого ядра, а те, в свою очередь, повышают активность тормозных ГАМК-ергических нейронов, за счет которых хвостатое ядро тормозит бледный шар. Выключение бледного шара приводит к растормаживанию мотонейронов и повышению тонуса мышц. В покое активируются дофаминергические нейроны черной субстанции. Они выделяют дофамин, который стимулирует постсинаптические D2-рецепторы, и тормозят тем самым холинергические нейроны хвостатого ядра. Кроме того, дофамин, выброшенный нейронами черной субстанции, стимулирует пресинаптические D1-рецепторы, которые располагаются на терминалях возбуждающих глутаматергических нейронов коры, и вызывает торможение этих нейронов (уменьшая тем самым их влияние на холинергические нейроны хвостатого ядра). В итоге, ГАМК-ергические нейроны хвостатого ядра остаются неактивными и прекращают оказывать тормозящее действие на бледный шар, он, в свою очередь, начинает тормозить a-мотонейроны спинного мозга, снижая тонус мышц. При паркинсонизме происходит гибель дофаминергических нейронов черной субстанции и тонус холинергических нейронов хвостатого ядра остается повышенным как при нагрузке, так и в покое. Глутаматергические нейроны коры постоянно стимулируют холинергические нейроны хвостатого ядра и те, в свою очередь, поддерживают ГАМК-ергические нейроны в активном состоянии. Таким образом, хвостатое ядро постоянно тормозит бледный шар и он не оказывает сдерживающего влияния на активность a-мотонейронов и тонус мышц остается повышенным как в покое, так и при нагрузке. Таким образом, при паркинсонизме имеет место дефицит дофаминергических влияний при усилении глутамат и холинергических влияний на тонус нейронов хвостатого ядра. Классификация противопаркинсонических средств: I. Средства, активирующие дофаминергические процессы: 1. Предшественники дофамина – леводопа; 2. Агонисты дофаминовых рецепторов – бромокриптин, перголид; 3. Средства, торомозящие метаболизм леводопы: A Ингибиторы ДОФА-декарбоксилазы – карбидопа, бензсеразид; B Ингибиторы МАО-В – селегилин; C Ингибиторы КОМТ – толкапон. 4. Средства, увеличивающие выделение дофамина – амантадин. II. Средства, блокирующие М,Н-холинорецепторы в ЦНС: тригексифенидил, бипериден.
Механизм действия: В ядрах ЦНС леводопа подвергается декарбоксилированию до дофамина, который восполняет собственный дефицит в нейронах черной субстанции экстрапирамидно системы. Воздействуя на D2-рецепторы холинергических нейронов хвостатого ядра, дофамин снижает их активность и уменьшает стимулирующее воздействие этих нейронов на ГАМК-ергические нейроны бледного шара. Растормаживание бледного шара способствует снижению тонической активности a-мотонейронов спинного мозга и снижению мышечного тонуса. ФК: После перорального введения леводопа всасывается в кишечнике активным транспортом при помощи переносчика для ароматических аминокислот. Богатая белком пища замедляет всасывание леводопы, т.к. ароматические аминокислоты конкурируют с ней за молекулы переносчика. Однако, процессу всасывания подвергается только около 10% введенной дозы, т.к. 90% лекарства разрушается до дофамина под воздействием ДОФА-декарбоксилазы кишечника. К сожалению, из всосавшихся 10% леводопы только 1-3% дозы поступает в нервную систему, а остальное подвергается разрушению ДОФА-декарбоксилазой крови и периферических тканей. При применении леводопы следует помнить, что витамин В6 (кофермент ДОФА-декарбоксилазы) усиливает периферический метаболизм леводопы и тем самым снижает ее активность.
Схема 2. Метаболизм леводопы: слева поступление леводопы в ЦНС в обычных условиях, справа – та же картина при блокаде активности ДОФА-декарбоксилаз периферических тканей. Метаболизм леводопы протекает при участии ДОФА-декарбоксилазы, которая переводит ее дафамин с последующим окислением ферментативными системами МАО и КОМТ до гомованилилминдальной кислоты. ФЭ: Леводопа уменьшает ригидность и олигокинезию у пациентов с болезнью Паркинсона, но чрезвычайно слабо влияет на тремор. Леводопа наиболее эффективна в первые несколько лет приема, однако, со временем ее эффект снижается, а количество нежелательных эффектов начинает увеличиваться. НЭ: При приеме леводопы нежелательные эффекты связаны в первую очередь с накоплением дофамина в ядрах ЦНС (центральные нежелательные эффекты) и периферических тканях (периферические эффекты). Периферические нежелательные эффекты: · Нарушения сердечного ритма, развитие приступов стенокардии, полиурия. Тахикардия и аритмия обусловлены активацией под влиянием дофамина b-адренорецепторов миокарда. Полиурия связана с расширением сосудов клубочков почек, которые несут дофаминовые D1-рецепторы, стимулируемые дофамином. · Анорексия, тошнота и рвота. Данные эффекты развиваются практически у каждого пациента, которому назначают даже минимальные дозы леводопы. По мере применения развивается привыкание и дозу в дальнейшем можно повышать до терапевтически значимой. Развитие рвоты связано со способностью дофамина стимулировать D1 и D5 рецепторы желудка, а также D2-рецепторы триггерной зоны рвотного центра продолговатого мозга. Триггерная зона располагается в области дна IV желудочка и лежит кнаружи от ГЭБ, поэтому ее возникновение связано не с концентрацией дофамина в ЦНС, а с его содержанием в крови и периферических тканях. Центральные нежелательные эффекты: · Ортостатическая гипотензия. Ортостатическая гипотензия возникает примерно у ⅓ пациентов, которые принимают леводопу и связана, как полагают, со способностью дофамина стимулировать адренорецепторы продолговатого мозга и снижать интенсивность симпатических сосудосуживающих влияний вазомоторных центров ЦНС. · Оральный гиперкинез – облизывание, оскаливание, почмокивание. · Хореические гиперкинезы – быстрые насильственные, неконтролируемые движения, обусловлены резким возрастанием концентрации дофамина, после приема леводопы. · Мышечные дистонии – внезапное застывание в аномальной позе, связано с падением концентрации дофамина перед очередным приемом лекарства. · Феномен «включения-выключения» или «on-off»-феномен – внезапные переходы от движения к полной неподвижности. · Тревога, бессонница, ночные кошмары – связаны с влиянием дофамина на систему ядер шва (гипногенная зона мозга). · Зрительные галлюцинации, бред, психоз – обусловлены стимуляцией D2-рецепторов лимбической системы. · Синдром «отмены» – возникает при внезапном прекращении приема леводопы, после длительного ее использования. Проявляется полной иммобилизацией, грубым тремором, злокачественной гипертермией, дыхательной и сердечной недостаточностью. Для купирования этого состояния необходимо внутривенное введение леводопы, апоморфина или бромокриптина. К достаточно редким нежелательным эффектам леводопы относят мидриаз и повышение внутриглазного давления (влияние на адренорецепторы радужки), гемолитическая анемия, гепато- и нефротоксичность, нарушения вкуса и обоняния, приапизм, окраска секретов (слюна, пот, слезная жидкость) в коричневый цвет. Режим дозирования: Лечение леводопой начинают с минимальных доз (120-250 мг 4-6 раз в день), которые постепенно увеличивают до создания оптимального эффекта. Увеличение дозы проводят 1 раз в 2-3 дня на 500-1000 мг. Как правило, поддерживающая доза при монотерапии леводопой составляет 1,5-8,0 г/сут. При возникновении нежелательных эффектов центрального типа («on-off»-феномен и др.) рекомендуется перейти на более частый прием дробных доз или использовать препараты леводопы продленного действия. При возникновении гиперкинезов иногда помогают «лекарственные каникулы» - отказ от приема леводопы на 3-21 дня. ФВ: таблетки по 0,25 Ингибиторы периферической ДОФА-декарбоксилазы. Карбидопа (Carbodopa), Бенсеразид (Benserazide). Эта группа лекарственных средств, которые сами по себе не оказывают терапевтического эффекта (противопаркинсонического действия).
При лечении паркинсонизма ингибиторы ДОФА-декарбоксилаз используют совместно с препаратами леводопы (эффект потенцирования). Т.о., на фоне применения ингибиторов можно вводить меньшие дозы леводопы. Как правило эффективная доза комбинированных препаратов леводопы и ингибиторов составляет 0,5-1,0 г/сут леводопы и 100-200 мг/сут ингибитора. Прекращение периферического декарбоксилирования леводопы снижает концентрацию дофамина в периферических тканях и уменьшает частоту и выраженность периферических нежелательных эффектов (гипотензии, тошноты, рвоты, полиурии). В то же время, усиление поступления леводопы в ЦНС чревато возрастанием частоты и выраженности центральных нежелательных эффектов (гиперкинезов, психозов, бессонницы и др.). ФВ: Синемет (Sinemet,) таблетки по 250 мг, содержащие 200 мг леводопы и 50 мг карбидопы; Мадопар (Madopar) капсулы по 125 и 250 мг, содержащие 100 и 200 мг леводоы соответственно в комбинации с 25 и 50 мг бенсеразида. Селегилин (Selegiline, Deprenyl, Niar). Механизм действия. Является избирательным ингибитором МАО типа B. В организме человека присуствует две изоформы фермента МАО: · МАО-А – располагается преимущественно на периферии (кишечник, печень, легкие) и в меньшей степени в ЦНС; проводит окислительное дезаминирование норадреналина, серотонина, дофамина, тирамина. · МАО-В – располагается преимущественно в ЦНС и в меньшей степени на периферии, проводит окислительное декарбоксилирование дофамина и тирамина.
ФЭ: Сам по себе селегилин оказывает лишь минимальный антипаркинсонический эффект. Это может быть связано с тем, что эндогенного дофамина у лиц страдающих паркинсонизмом недостаточно. Однако, в комбинации с препаратами леводопы селегилин позволяет усилить ее эффект. Показано, что селегилин замедляет прогрессирование заболевания. Возможно, что это связано с его антиоксидантным эффектом и способностью защищать дофаминергические нейроны черной субстанции от повреждающего действия свободных радикалов. К сожалению, через 1-2 года терапевтический эффект селегилина заметно ослабевает. Показания и режим дозирования: Лечение паркинсонизма у пациентов с ухудшающимся ответом на терапию леводопой. Селегилин назначают внутрь в 2 приема по 5 мг утром и в полдник. НЭ: Подобно ингибиторам периферической ДОФА-декарбоксилазы селегилин увеличивает частоту и выраженность центральных нежелательных эффектов леводопы. В процессе метаболизма селегилина образуется амфетамин, который оказывает выраженное стимулирующее влияние на ЦНС, вызывая возбуждение, спутанность сознания, параноидные психотические реакции, повышение артериального давления. Поскольку селегилин не нарушает активность МАО-А он не препятствует разрушению других аминов и не вызывает гипертонической реакции при употреблении в пищу продуктов, богатых этими аминами. Применение с аналогичными целями неселективных ингибиторов МАО-А и В неприемлемо. При этом нарушается дезаминирование не только дофамина, но и других биогенных аминов (тирамина, серотонина), которые содержатся в ряде продуктов питания, технология производства которых связана с процессами ферментации. Употребление в пищу этих продуктов приводило бы у таких пациентов к развитию гипертонических кризов по типу «сырного» и «серотонинового» синдромов, описанных у лиц, которые принимают ингибиторы МАО для лечения депрессии. ФВ: таблетки по 5 и 10 мг.
Показания и режим дозирования: Толкапон дополняет антипаркинсонический эффект других лекарственных средств, тормозящих метаболизм леводопы. Чаще всего его применяют в сочетании с селегилином, ингибиторами ДОФА-декарбоксилаз в дополнение к терапии леводопой у пациентов, которые плохо переносят монотерапию леводопой или недостаточно отвечают на нее. Толкапон применяют внутрь по 100-200 мг 3 раза в день во время еды. НЭ: Наиболее частым нежелательным эффектом является диарея, которая развивается через 2-4 месяца регулярного применения толкапона и связана с усилением перистальтики кишечника. Толкапон увеличивает частоту и выраженность центральных нежелательных эффектов леводопы, оказывает гепатотоксическое действие. Справедливости ради, следует отметить, что гепатотоксическое действие толкапона привело к запрещению его применения в ряде европейских стран. В настоящее время проводятся интенсивные клинические испытания менее токсичных ингибиторов КОМТ – энтокапона и нитекапона. ФВ: таблетки по 100 и 200 мг. Бромокриптин (Bromocriptin, Parlodel). Производное алкалоидов спорыньи. Механизм действия: Бромокриптин является агонистом постсинаптических D2-дофаминовых рецепторов.
ФЭ: · Противопаркинсонический эффект. Бромокриптин активирует D2-рецепторы нейронов хвостатого ядра и воспроизводит тем самым тормозящий эффект, который оказывают нейроны черной субстанции или леводопа на хвостатое ядро. Торможение хвостатого ядра приводит к активации нейронов бледного шара и усилению его нисходящих тормозных влияний на a-мотонейроны спинного мозга. В итоге, бромокриптин снижает брадикинезию и ригидность. · Эндокринологические эффекты. Бромокриптин вызывает угнетение секреции пролактина. Этот эффект связан с влиянием на рецепторы гипофиза. В настоящее время полагают, что гипоталамический гормон пролактостатин является ни чем иным, как дофамином. Бромокриптин, стимулируя D2-дофаминовые рецепторы гипофиза, тормозит образование и секрецию пролактина. Кроме того, показано, что бромокриптин (как и другие дофаминомиметики) нормализует уровень гормона роста: снижает патологически повышенную его секрецию и несколько повышает нормальную секрецию СТГ. Показания к применению: 1. Лечение гиперпролактинемии и связанных с ней нарушений менструального цикла, женского и мужского бесплодия, лечение пролактином (гормонпродуцирующих опухолей гипофиза, выделяющих пролактин). Бромокриптин назначают в дозе 7,5-20 мг/сут. 2. Подавление лактации в послеродовом периоде (т.к. синтез и секреция молока определяются уровнем пролактина). Используют бромокриптин в дозах 7,5-15 мг/сут. 3. Лечение акромегалии (обусловленой гормонпродуцирующими опухолями гипофиза). Эффективные дозы составляют 1,25-5,0 мг/сут. 4. Лечение диффузной формы кистозно-фиброзной мастопатии. Показано, что бромокриптин снижает число кист и узлов в тканях молочной железы, полагают, что это связано с нормализацией секреции пролактина и соотношения эстрогенов и прогестерона. Эффективные дозы составляют 5,0-10,0 мг/сут. 5. Лечение паркинсонизма. Используют дозы 10-40 мг/сут. Режим дозирования: Во всех случаях подбор оптимальной дозы бромокриптина проводят по следующей схеме. В 1-ую неделю его принимают внутрь по 1,25 мг на ночь, в течение второй недели – по 2,5 мг на ночь, с 3-ей недели – по 2,5 мг 2 раза в день и с 4-ой недели – по 2,5 мг 3 раза в день (что соответствует 7,5 мг/сут). При необходимости дальнейшего увеличения его проводят каждые 3 дня повышая дозу на 2,5 мг. НЭ: · Со стороны ЖКТ отмечаются эффекты характерные для леводопы и других дофаминомиметиков – анорексия, тошнота и рвота. · Ортостатический коллапс при первых приемах. · Эритромелалгия – болезненный спазм сосудов пальцев рук, который сменяется их внезапным расширением с гиперемией, отеком и жжением. Провоцирующим фактором является контакт с водой. · Эрготизм – сочетание спазмов сосудов конечностей, внутренних органов с изменениями психики в виде галлюцинаций и бреда. ФВ: таблетки по 0,0025; 0,004 и 0,01; капсулы по 0,004 и 0,01; раствор для приема внутрь 4 мг/5 мл (0,08%) во флак. по 100 мл
В отличие от бромокриптина перголид не подвержен пресистемной элиминации, имеет более длительный период полувыведения (около 27 часов), что позволяет назначать его 1 раз в день. Кроме того, он более доступен по цене, что немаловажно, если учитывать чрезвычайно высокую стоимость терапии бромокриптином. ФЭ: Для перголида характерны те же эффекты, что и для бромокриптина. Показания для применения и режим дозирования. В настоящее время одобрено применение перголида только для лечения паркинсонизма. Прием перголида начинают с 0,05 мг на ночь в течение 2 дней, затем дозу повышают на 0,1 мг каждые 3 дня. Как правило, эффективными являются дозы 0,4-0,5 мг/сут, однако, при необходимости можно увеличивать их до 3-5 мг/сут. При плохой переносимости можно принимать перголид в 2-3 приема. НЭ: Перголид лучше переносится, чем бромокриптин и реже вызывает ортостатический коллапс и рвоту. При длительном применении перголида возможно появление плеврального или перикардиального выпота, фиброза плевральной полости или забрюшинного пространства. Возможно развитие психотических реакций. ФВ: таблетки по 0,00005; 0,00025 и 0,001 Амантадин (Amantadine, Midantan). Первоначально был создан как противовирусное средство из группы адамантановых производных. Его противопаркинсоническая активность была обнаружена случайно.
· Блокада NMDA-рецепторов на поверхности холинергических нейронов хвостатого ядра. В результате это блокады не происходит стимулирующего воздействия на эти клетки возбуждающих глутаматергических нейронов коры и активность холинергических нейронов понижается. При этом уменьшается активность связанных с ними ГАМК-ергических нейронов хвостатого ядра и они прекращают тормозить нейроны бледного шара. В итоге, ГАМК-ергические нейроны бледного шара снижают тонус a-мотонейронов спинного мозга и тонус скелетных мышц. · Полагают, что амантадин может усиливать выделение дофамина в синаптическую щель из нейронов черной субстанции и тормозить его обратный нейрональный захват. В итоге, в области нейронов хвостатого ядра концентрация дофамина повышается и он оказывает тормозящее действие на холинергические нейроны, дополняя эффект амантадина, связанный с блокадой NMDA-рецепторов. · Амантадин обладает слабой М-холиноблокирующей активностью. Поэтому он блокирует М-холинорецепторы тормозных нейронов хвостатого ядра и препятствует их активации ацетилхолином, который выделяется возбуждающими холинергическими нейронами. ФЭ: Амантадин оказывает противопаркинсоническое действие и уменьшает выраженность акинезии, ригидности и тремора в равной мере, хотя и с меньшей эффективностью, чем леводопа. По эффективности в 15-20 раз слабее леводопы. Чаще всего амантадин используют как вспомогательное средство при лечении леводопой (для усиления ее эффекта). Терапевтический эффект леводопы развивается через 3-5 дней регулярного приема, но, к сожалению, уже через несколько недель терапии он значительно ослабевает. Благодаря противовирусному эффекту в отношении вируса гриппа типа А амантадин используют для лечения и профилактики гриппа. Режим дозирования: Как противопаркинсоническое средство амантадин применяют по 100-200 мг 2 раза в день внутрь. НЭ: Чаще всего наблюдаются изменения со стороны ЦНС в виде депрессии, бессонницы, психомоторного возбуждения, галлюцинаций. В высоких (более 200 мг/сут) дозах способен вызвать судороги. При применении амантадина возможно развитие периферических отеков и livedo reticularis – зигзагообразной пигментации кожи в виде «стрел молний» (чаще всего возникает в области лодыжек). Как правило эти симптомы исчезают через 1 месяц после регулярного применения амантадина. М-холиноблокирующее действие амантадина может приводить к появлению сухости во рту, глотке, запорам и задержке мочи (особенно у пожилых мужчин). ФВ: таблетки по 0,1 и 0,2.
ФЭ: Тригексифенидил оказывает противопаркинсоническое действие и устраняет главным образом тремор, в меньшей степени воздействуя на акинезию и ригидность. Достаточно эффективно подавляет синдром вегетативных расстройств, который имеет место у таких больных (устраняет слюнотечение, потливость и др.). Показания к применению: Учитывая чрезвычайно низкую эффективность этой группы лекарств (эффект возникает только у 10-30% пациентов), достаточно быстрое развитие привыкания и потенциально высокую токсичность данные средства используют в настоящее время достаточно редко, в основном как лекарства, дополняющие леводопу при выраженном треморе или при лечении паркинсонизма, который вызван приемом антипсихотических средств, блокирующих дофаминовые рецепторы (нейролептики). Режим дозирования: Лечение тригексифенидилом начинают с минимальной дозы 1-2 мг/сут, которую постепенно повышают до максимально переносимой (определяют по возникновению нежелательных эффектов). Как правило, эффективные дозы тригексифенидила составляют 6-20 мг/сут НЭ: Чаще всего наблюдаются изменения со стороны ЦНС в виде сонливости, замедления мышления, нарушения внимания, необъяснимых колебаний настроения, ярких красочных галлюцинаций и иллюзорного восприятия мира. Чаще всего эти эффекты воникают через несколько дней после применения тригексифенидила. Несмотря на центральное М-холиноблокирующее действие, возможно появление периферических эффектов, связанных с блокадой М-холинорецепторов – сухости во рту, глотке, рези в глазах, нарушения аккомодации и светобоязни, повышения внутриглазного давления, тахикардии, запора, задержки мочи. ФВ: таблетки по 0,001; 0,002 и 0,005.
При лечении паркинсонизма бипериденом средние ориентировочные дозы составляют 9-12 мг/сут внутрь. Иногда используют внутривенное введение биперидена (5 мг) для купирования острых тяжелых дистонических реакций, возникающих при лечении леводопой. ФВ: таблетки 0,002 и пролонг. в оболочке 0,004; раствор 0,5% в амп. по 1 мл. Таблица 1. Влияние лекарственных веществ на кардинальные симптомы паркинсонизма.
ПРОТИВОЭПИЛЕПТИЧЕСКИЕ И ПРОТИВОСУДОРОЖНЫЕ СРЕДСТВА Способствовал тому страшный эпилептический вопль Смердякова, лежавшего в соседней комнатке без сознания, – тот вопль, которым всегда начинались его припадки падучей…Зажгли огонь и увидали, что Смердяков все еще не унимается и бьется в своей каморке, скосил глаза, а с губ его текла пена. Ф.М. Достоевский «Братья Карамазовы» Эпилепсия – заболевание, которое характеризуется периодическим возникновением судорожного синдрома, приводящего с течением времени к изменению психики больного человека. Возникновение судорог при эпилепсии обусловлено наличием в головном мозге эпилептогенного очага. Этот очаг представляет собой группу нейронов, мембраны которых обладают высокой спонтанной проницаемостью для ионов натрия и кальция. Благодаря току ионов внутрь клеток мембраны нейронов спонтанно деполяризуются и гененируют гиперсинхронные импульсы, способные не только распространяться по всей коре головного мозга, но и подавлять активность нормальных нейронов. Противоэпилептическими называют лекарственные средства, которые снижают частоту и выраженность судорожных припадков при эпилепсии. Следует особо отметить, что противоэпилептические средства не назначают для купирования уже развившихся припадков (за исключением эпилептического статуса), их применяют исключительно с целью профилактики приступов судорог у больного человека. Кроме того, будет неверным утверждать, что противоэпилептические средства излечивают эпилепсию, они лишь позволяют сдержать развитие заболевания или даже приостановить его, но не способны полностью устранить эпилепсию. Классификация и характеристика различных форм эпилепсии. До настоящего времени не существует единой общепринятой классификации эпилепсии. С фармакотерапевтических позиций удобно пользоваться следующей упрощенной классификацией эпилептических припадков: A. Генерализованные судорожные припадки – припадки судорог, при которых эпилептогенный очаг охватывает всю кору головного мозга. 1. большие судорожные припадки (grand mal); 2. абсансы (petit mal); 3. миоклонус-эпилепсия; 4. фебрильные припадки (судорожные припадки, обусловленные гипертермией, как правило развиваются у детей от 3 мес до 5 лет). B. Парциальные (фокальные) судорожные припадки – припадки судорог, при которых эпилептогенный очаг охватывает четко ограниченную область коры головного мозга. 1. простые парциальные припадки; 2. сложные парциальные припадки (психомоторные эквиваленты). C. Неонатальные припадки – припадки судорог, которые развиваются у детей первого года жизни. Большие судорожные припадки являются одной из самых распространенных и, одновременно, самых тяжелых форм эпилепсии. Развитию большого судорожного припадка обычно предшествует аура – особое психическое состояние с появлением «предвестников» приступа. Во время ауры пациенты могут испытывать сенсорные иллюзии (ощущать «могильный» запах, дуновение ветра, «пророческие видения», изменение окраски предметов и т.п.). После ауры человек теряет сознание и у него развиваются вначале тонические, а затем клонические судороги всех мышц. Припадок завершается часто непроизвольным мочеиспусканием и дефекацией, после чего возникает длительный послеприпадочный сон. После припадка возникает ретроградная амнезия (пациент не помнит событий предшествующих припадку). Абсансы чаще всего возникают в детстве (3-5 лет) и продолжаются с той или иной частотой до пубертаного периода. Типичная картина абсанса характеризуется внезапной потерей сознания, при которой пациент как бы выпадает из окружающей реальности – он на несколько секунд или минут застывает, иногда это сопровождается быстрым морганием, почмокиванием, пропульсивными движениями (покачивание, кивание и т.п.), затем также внезапно человек приходит в сознание. При этом сам пациент не осознает своего временного отключения и продолжает выполнять ту работу (беседу, письмо, еду и т.п.) за которой его застиг припадок. Миоклонус-эпилепсия является одной из самых неблагоприятных форм эпилепсии, она плохо поддается медикаментозной коррекции и быстро приводит к формированию дефекта личности. Чаще всего эта форма эпилепсии начинается после органического поражения головного мозга – энцефалита, отравления нейротропными ядами, уремии или гипоксии. Миоклонус характеризуется внезапным возникновением частых подергиваний групп мышц при полном сохранении сознания, обычно миоклонический припадок может многократно повторяться с интервалами в несколько минут. Простые парциальные судороги характеризуются возникновением приступа судорог в группе мышц, которая соответствует локализации эпилептогенного очага (судороги одной руки, судороги пальца и т.п.). Часто эпизоду судорог предшествует аура. Эти припадки протекают при сохранении сознания. Сложные парциальные припадки характеризуются отсутствием типичной судорожной картины как таковой. У пациента наблюдается сумеречное расстройство сознания с возникновением галлюцинаций, нарушением мышления, возникновением психомоторного автоматизма (выполнение сложных целенаправленных движений и поступков без контроля со стороны сознания). Как правило, после выхода из припадка у человека наблюдается антероградная амнезия на события, которые с ним происходили во время припадка. Отдельно выделяют эпилептический статус – особое состояние, при котором судорожные припадки следуют друг за другом, при этом завершение одного припадка происходит в тот момент, когда начинается следующий. Основным критерием эпилептического статуса является отсутствие даже кратковременного периода возвращения пациента в нормальное состояние между припадками. Сравнительная характеристика различных типов припадков показана в таблице 1. Таблица 1. Сравнительная характеристика судорожных припадков при эпилепсии.
Классификация противоэпилептических средств. До настоящего времени нет общепринятой классификации данной группы лекарственных средств. Наиболее удобной является химико-фармакологическая классификация, которая учитывает строение и механизм действия противоэпилептических средств. Согласно этой классификации можно выделить 4 группы противоэпилептических средств. A. Средства, угнетающие активность Na+-каналов мембран нейронов: 1. производные гидантоина – фенитоин; 2. производные иминостильбена – карбамазепин. B. Средства, угнетающие активность Ca2+-каналов T- и L-типов: 1. производные сукцинимидов – этосуксимид. C. Средства, повышающие активность ГАМК-ергической системы: 1. производные барбитуровой кислоты – фенобарбитал; 2. производные жирных кислот – натрия и магния вальпроат; 3. синтетические аналоги ГАМК – габапентин; 4. производные бензодиазепинов – клоназепам, диазепам. D. Средства, угнетающие активность глутаматергической системы: 1. производные фенилтриазинов – ламотриджин.
Фенитоин (Phenytoin, Diphenin, Dylantine). Является производным гидантоина. Был синтезирован в 1908 г как снотворное средство, аналог барбитуратов. Однако, он не нашел практического применения, так как практически не обладал седативным и снотворным эффектом. В 1938 г в процессе скриннинга противоэпилептических средств была обнаружена его противосудорожная активность.
Кроме того, установлено, что фенитоин обладает также рядом других механизмов действия:
· фенитоин нарушает ток ионов кальция в клетку по кальциевым каналам, это нарушает процесс деполяризации и генерации длительных возбуждающих потенциалов, необходимых для образования гиперсинхронных пачек нервных импульсов. · В высоких концентрациях фенитоин подавляет процесс выделения в синапс норадреналина и серотонина и нарушает обратный нейрональный захват дофамина. В конечном итоге, это приводит к затруднению передачи в моноаминергических синапсах ЦНС. ФК: после перорального приема фенитоин хорошо и полностью всасывается, максимальная концентрация фенитоина в плазме крови возникает через 4-12 ч. Однако, следует помнить, что скорость и полнота абсорбции фенитоина отличаются у таблеток, произведенных разными фармацевтическими компаниями, поэтому самостоятельная замена одного препарата фенитоина на другой (даже аналогичной дозировки) без контроля врача недопустима. Возможно внутривенное введение натриевой соли (phenytoin sodium) или фосфорного эфира (fosphenytoin) фенитоина, при этом следует помнить, что нельзя растворять фенитоин в растворах глюкозы, так как возможно его окисление. Внутримышечное введение фенитоина недопустимо, поскольку в мышцах фенитоин образует плохорастворимые преципитатыи всасывается медленно и неполно (при внутримышечном введении биодоступность фенитоина меньше, чем при его пероральном введении). В крови фенитоин на 90% связан с белками крови и уровень свободной фракции составляет около 10%. В том случае, если человек одновременно принимает другие средства, которые также способны интенсивно связываться с белком (сульфаниламидные препараты, сахароснижающие средства, нестероидные противовоспалительные средства, оральные антикоагулянты и др.), происходит конкуренция за белки крови и часть фенитоина может переходить в свободное состояние. Вытеснение даже 10% фенитоина из связи с белком приводит к тому, что уровень его свободной фракции увеличивается в 2 раза (возрастание с 10% до 20%) и соответственно в 2 раза возрастает риск нежелательных эффектов и интоксикации. Для фенитоина характерна нелинейная кинетика – в малых дозах скорость его элиминации пропорциональна введенной дозе и концентрация фенитоина постепенно стабилизируется на некотором стационарном уровне (кинетика первого порядка), но как только величина дозы фенитоина превысит метаболические возможности ферментов печени, кинетика фенитоина переходит в нулевой порядок, т.е. скорость элиминации остается постоянной и не зависит от дозы. Поэтому даже при незначительном увеличении дозы возможен резкий скачок концентрации фенитоина, который приводит к развитию отравления. Биотрансформация фенитоина происходит в печени путем глукуронидации и гидроксилирования (при участии цитохромов Р450). В процессе лечения фенитоином происходит индукция (повышение активности) этих ферментов, что может привести к увеличению разрушения как самого фенитоина, так и других лекарств, принимаемых совместно с ним. ФЭ и показания к применению: 1. Фенитоин оказывает противосудорожное действие при парциальных эпилептических припадках, grand mal; при этом в терапевтических дозах он практически не влияет на когнитивно-мнестические процессы, т.к. не изменяет функции нормальных нейронов. 2. Фенитоин способен купировать судороги при эпилептическом статусе, при этом обычно используют внутривенное введение натриевой соли или фосфорного эфира фенитоина, реже прибегают к ректальному введению при помощи микроклизм. 3. Фенитоин устраняет боль при невралгии тройничного нерва (заболевание, при котором даже минимальное раздражение нерва – например дуновение ветра в лицо, вызывает тяжелый приступ болей). Данный эффект нельзя назвать анальгетическим, поскольку проявляется он только при невралгии и не возникает при любых других видах болей. 4. Фенитоин оказывает противоаримическое действие, которое связано с его способностью блокировать натриевые каналы мембран кардиомиоцитов (мембраностабилизирующее действие). В отличие от всех других противоаритмических средств, фенитоин не оказывает никакого влияния на проводящую систему миокарда, поэтому он особенно эффективен при аритмиях, вызванных передозировкой сердечных гликозидов (при этих аритмиях повышается возбудимость только рабочего миокарда, а возбудимость проводящей системы не изменяется или даже понижается). РД: Ввиду того, что кинетика фенитоина нелинейна, подбор и изменение дозы фенитоина следует выполнять крайне осторожно. Вначале назначают нагрузочную дозу фенитоина – с интервалом в 2 часа пациент должен принять вначале 3 таблетки, затем еще раз 3 таблетки и наконец 2 таблетки фенитоина. Таким образом, всего за 4 часа он принимает 8 таблеток фенитоина. После этого пациента переводят на поддерживающую дозу фенитоина. Он принимает по 3 таблетки фенитоина 1 раз в день (как правило утром). При необходимости дозу фенитоина повышают на ¼ таблетки каждую неделю. Максимальная возможная поддерживающая доза 4 таблетки в день. Детям фенитоин назначают в дозе 5 мг/кг веса в день. При лечении фенитоином следует стремиться поддерживать его концентрацию на терапевтическом уровне. О концентрации фенитоина в крови судят либо по лабораторным данным мониторирования концентрации, либо косвенно, по сопутствующим каждому уровню клиническим симптомам: Таблица 2. Клинические симптомы при различных уровнях фенитоина в крови.
При использовании фенитоина в качестве противоаритмического средства его вводят внутривенно медленно в дозе 4 мг/кг веса под контролем ЭКГ. НЭ: Различают нежелательные эффекты фенитоина, которые проявляются при низком и при высоком уровне лекарства. · При низком уровне лекарства: ♦ гипертрофия десен (возникает в 20% случаев) – чаще появляется у молодых лиц, выраженность ее может быть уменьшена при соблюдении гигиены полости рта; ♦ гирсутизм, огрубение черт лица, акне; ♦ реакции гиперчувсвительности – сыпи, лимфаденопатия, волчаночный синдром; ♦ гематотоксические реакции – нейтропения, мегалобластная анемия (обусловлена увеличением выведения и снижением абсорбции витамина Вс); ♦ остеопороз и остеомаляция (снижение чувствительности периферических тканей к витамину D, нарушение обмена кальция); ♦ снижение выделения инсулина поджелудочной железой (гипергликемические состояния); ♦ прием фенитоина во время беременности приводит к развитию гидантоинового синдрома плода – сочетания микроцефалии с расщеплением губы, незаращенным небом, гипоплазией ногтей и фаланг пальцев. Этот эффект обусловлен образованием ареноуксусного метаболита фенитоина; ♦ толерантность (привыкание) к фенитоину и снижение эффекта других лекарственных средств на фоне фенитоина, что обусловлено индукцией микросомальных ферментов при его применении (особого внимания требуют пациентки, принимающие оральные контрацептивы, т.к. у них необходимо увеличить дозу контрацептивов); ♦ синдром рикошета – возникает при внезапном прекращении применения фенитоина, характеризуется резким возрастанием числа судорожных эпизодов у пациента. · При высоком уровне лекарства: ♦ нейротоксические реакции – атаксия, головокружение, диплопия. Нистагм, сонливость, нарушение поведения, галлюцинации, судороги; ♦ тошнота и рвота (эти эффекты можно уменьшить, если принимать высокие дозы фенитоина во время еды); ♦ при внутривенных инъекциях возможно локальное повреждение сосудов с развитием отека и обесцвечивания кожи по ходу вены, снижение артериального давления, развитие аритмии. ФВ: табл. 0,117
МД: Трехмерная конфигурация карбамазепина весьма близка к конформации фенитоина, поэтому он также способен связываться с инактивированными натриевыми каналами и пролонгировать состояние их инактивации, нарушая поступление ионов натрия в клетку и генерацию ПД. Относительно недавно было установлено, что карбамазепин является антагонистом NMDA-рецепторов для глютаминовой кислоты. Карбамазепин взаимодействует с активным центром этих рецепторов и препятствует их активации глютаминовой кислотой. Поскольку рецепторы остаются неактивными не происходит открытия сопряженного с ними кальциевого канала и нарушается поступление ионов кальция в клетку. Поступление кальция в пресинаптическое окончание необходимо для выделения медиаторов, а поступление этих ионов в постсинаптическое окончание – для генерации длительных возбуждающих потенциалов. Оба этих процесса под влиянием карбамазепина нарушаются. Кроме того, поступление избытка ионов кальция в нейрон во время судорожной активности приводит к перегрузке ионами кальция митохондрий и их гибели. Карбамазепин оказывает защитный эффект и уменьшает тем самым дегенеративные процессы в нейронах под влиянием избытка ионов кальция. ФК: Карбамазепин хорошо всасывается после перорального применения, с белками крови связывается на 60-70%, но практически не вытесняется другими лекарственными средствами из связи с белком и поэтому совместно с другими лекарствами может применяться без дополнительной коррекции дозы. При регулярном использовании карбамазепин вызывает индукцию микросомальных ферментов печени и усиливает свой собственный метаболизм. Поэтому с течением времени скорость выведения карбамазепина возрастает (вначале его период полуэлиминации составляет около 36 часов, а затем снижается до 20 часов).
В процессе метаболизма карбамазепина образуется 10,11-эпоксикарбамазепин, который обладает собственной противосудорожной активностью, составляющей»30% активности карбамазепина. ФЭ и показания к применению: 1. Противосудорожный эффект. Карбамазепин является средством выбора при лечении сложных парциальных припадков (психомоторных эквивалентов), кроме того, он эффективен при простых парциальных припадках и grand mal. 2. Карбамазепин устраняет боль при невралгии тройничного нерва. Эффект карбамазепина превосходит эффект фенитоина и является весьма специфичным именно для этого заболевания, поэтому иногда карбамазепин применяют с диагностической целью при боли в области лица неясного генеза (если карбамазепин снимает эту боль, то диагноз невралгии считается подтвержденным). 3. Карбамазепин обладает способностью «стабилизировать» настроение, что используется при маниакальной стадии маниакально-депрессивного психоза как альтернатива солям лития. РД: Карбамазепин применяют исключительно перорально в дозе 1,0-2,0 г/сут, разделенной на 3-4 приема. Детям карбамазепин назначают в дозе 15-25 мг/кг/сут. НЭ: 1. Диплопия – для нее характерна строгая периодичность, двоение в глазах возникает в определенное время суток и продолжается около часа. Часто ее можно устранить просто изменив время приема препарата. 2. Атаксия. 3. Сонливость, вялость, головокружение. 4. Отеки и гипонатриемия (особенно выражены у пожилых людей). Полагают, что отеки связаны со способностью карбамазепина усиливать секрецию АДГ. 5. Тошнота, диарея, усугубление судорог. 6. Снижение эффективности комбинированных оральных контрацептивов, вследствие индукции микросомальных ферментов печени. 7. Апластическая анемия и агранулоцитоз – чаще всего эти эффекты возникают у пожилых лиц, которые принимают карбамазепин в связи с невралгией тройничного нерва. 8. Идиосинкразия – протекает в форме эритемы кожи и фотосенсибилизации. ФВ: табл. 0.1 и 0,2; таблетки ретард по 0,2 и 0,4.
Показано, что в высоких дозах этосуксимид ингибирует Na+,K+-АТФазу и нарушает тем самым процесс реполяризации мембраны, продлевая состояние сниженной возбудимости нейронов. В высоких дозах этосуксимид ингибирует активность ГАМК-транаминазы (ГАМК-Т), фермента, который разрушает ГАМК до янтарного полуальдегида, поэтому в синапсах ЦНС возрастает количество ГАМК, которая оказывает тормозящее влияние на нейроны. ФК: Этосуксимид практически полностью реабсорбируется из ЖКТ, практически не свяхывается с белками крови, поэтому прием других лекарств не оказывает никакого влияния на его уровень в крови. Метаболизм этосуксимида происходит в печени с образованием гироксипроизводного под влиянием ферментов системы цитохрома Р450. Выведение этосуксимида крайне медленное, период полуэлиминации составляет около 40 часов (по некоторым данным до 72 часов), т.е. полное выведение этосуксимида после однократного применения занимает около 2 недель. ФЭ: Этосуксимид является лекарственным средством с очень узким спектром активности. Он оказывает эффект только при абсансах. Для достижения терапевтического эффекта требуется прием 750-1500 мг этосуксимида в день. Поскольку период полуэлиминации этосуксимида достаточно продолжительный, его можно было бы применять 1 раз в день, однако этому мешает мощное раздражающее влияние этого лекарства на слизистые оболочки ЖКТ (боли в желудке, тошнота, рвота), поэтому дозу этосуксимида обычно применяют в 2 приема. НЭ: Наиболее значимым является раздражающее действие этосуксимида на слизистые оболочки ЖКТ (его можно уменьшить, если начинать прием этосуксимида с малых доз и делить суточную дозу на 2 приема). Реже возможны преходящая сонливость, усталость. ФВ: капс. 250 мг
Механизм действия: Фенобарбитал активирует барбитуровый участок алостерического центра ГАМКА-хлоридного ионофорного комплекса, который включает в себя хлидный канал, ГАМКА-рецептор и 2 аллостерических центра – барбитуровый и бензодиазепиновый. Активация барбитурового центра комплекса приводит к резкому повышению сродства ГАМКА-рецепторов к ГАМК, при этом даже ничтожные количества ГАМК могут активировать рецептор и открыть хлоридный канал, при этом под влиянием фенобарбитала длительность пребывания канала в открытом состоянии также резко увеличивается. Ионы хлора поступая в клетку через открытый канал вызывают гиперполяризацию мембраны нейрона эпилептогенного очага и снижают его возбудимость. Показано, что фенобарбитал блокирует АМРА-рецепторы для глютаминовой кислоты. АМРА-рецепторы связаны с натриевыми каналами мембраны. Их блокада приводит к тому, что глютамат не способен активировать рецептор и каналы остаются закрытыми. Прекращается поступление ионов натрия в клетку и генерация ПД. Кроме того, поступление избыточных количеств натрия в нейроны при судорожном припадке способствует развитию отека клетки, т.к. по осмотическому градиенту вслед за натрием в клетку поступает вода. Поскольку фенобарбитал снижает поступление натрия в нейрон, он косвенно способствует уменьшению отека нервной ткани. В чрезвычайно больших количествах (которые не создаются в организме при обычной противосудорожной терапии) фенобарбитал способен блокировать Na+- и Са2+-каналы (L- и N-типов), что также снижает возможности генерации клеткой возбуждающих потенциалов. ФК: После перорального применения всасывается медленно. Фенобарбитал является слабой кислотой с рКа=7,3 поэтому при рН крови около 50% принятого лекарства находится в ионизированном состоянии и в ЦНС способно проникать только ½ принятой дозы. Во время судорог развивается гипоксия, которая приводит к ацидозу и доля неинонизированного фенобарбитала в крови повышается, а следовательно увеличивается его поступление в ЦНС и усиливается противосудорожный эффект лекарства.
Метаболизм фенобарбитала протекает в печени при участии цитохромов Р450. В процессе метаболизма происходит индукция (увеличение активности) микросомальных ферментов печени и ряда других ферментных систем гепатоцитов (аланиновой синтетаза, g-глютамил трансаминазы, глюкуронидазы и др.). Поэтому эффект ряда лекарственных средств на фоне приема фенобарбитала ослабляется (это обусловлено увеличением из разрушения). Выводится фенобарбитал крайне медленно, период полуэлиминации для него составляет 4-5 дней, поэтому для проведения адекватной противосудорожной терапии достаточно однократного применения фенобарбитала в сутки. Элиминация фенобарбитала осуществляется почками, при этом 50% введенного лекарства выводится в неизмененном виде. ФЭ: Дата добавления: 2015-01-12 | Просмотры: 1236 | Нарушение авторских прав |
 Леводопа (Levodopa, Dopar). Леводопа – левовращающий изомер диоксифенилаланина, используется для восполнения дефицита дофамина в ядрах экстрапирамидной системы. Поскольку ГЭБ непроницаем для медиаторов (в том числе и дофамина), применение для лечения паркинсонизма самого дофамина бессмыслено и не позволяет добиться терапевтического эффекта. Леводопа будучи предшественником дофамина способна проникать через ГЭБ.
Леводопа (Levodopa, Dopar). Леводопа – левовращающий изомер диоксифенилаланина, используется для восполнения дефицита дофамина в ядрах экстрапирамидной системы. Поскольку ГЭБ непроницаем для медиаторов (в том числе и дофамина), применение для лечения паркинсонизма самого дофамина бессмыслено и не позволяет добиться терапевтического эффекта. Леводопа будучи предшественником дофамина способна проникать через ГЭБ.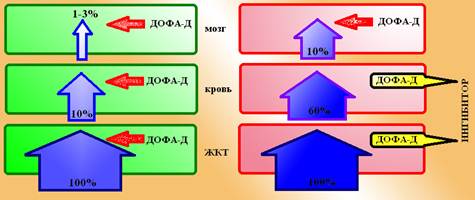
 Механизм действия: Ингибиторы ДОФА-декарбоксилазы не проникают через ГЭБ. Они конкурируют с леводопой в периферических тканях за активный центр фермента и, связываясь с ним, препятствуют декарбоксилированию леводопы в дофамин. Таким образом, на периферии (кишечник, печень, легкие) разрушение леводопы прекращается и бóльшая часть лекарственного вещества (около 10%) достигает ЦНС. В то же время, активность ДОФА-декарбоксилазы ЦНС не нарушается и леводопа без помех превращается в дофамин, который оказывает свое терапевтическое действие.
Механизм действия: Ингибиторы ДОФА-декарбоксилазы не проникают через ГЭБ. Они конкурируют с леводопой в периферических тканях за активный центр фермента и, связываясь с ним, препятствуют декарбоксилированию леводопы в дофамин. Таким образом, на периферии (кишечник, печень, легкие) разрушение леводопы прекращается и бóльшая часть лекарственного вещества (около 10%) достигает ЦНС. В то же время, активность ДОФА-декарбоксилазы ЦНС не нарушается и леводопа без помех превращается в дофамин, который оказывает свое терапевтическое действие. В дозах менее 10 мг/сут селегилин связывается практически исключительно с активным центром МАО-В и нарушает способность этого фермента разрушать дафамин. Следовательно, на фоне селегилина эффект леводопы усиливается и продлевается. Это объясняется тем, что замедляется разрушение дофамина, который образуется из леводопы.
В дозах менее 10 мг/сут селегилин связывается практически исключительно с активным центром МАО-В и нарушает способность этого фермента разрушать дафамин. Следовательно, на фоне селегилина эффект леводопы усиливается и продлевается. Это объясняется тем, что замедляется разрушение дофамина, который образуется из леводопы. Толкапон (Tolcapone, Tasmar). МД: Является обратимым ингибитором КОМТ. При приеме внутрь, толкапон хорошо всасывается и проникает через ГЭБ в ЦНС. Связываясь с активным центром КОМТ он препятствует метилированию катехоламинов (в том числе и дофамина) и скорость метаболизма дофамина замедляется.
Толкапон (Tolcapone, Tasmar). МД: Является обратимым ингибитором КОМТ. При приеме внутрь, толкапон хорошо всасывается и проникает через ГЭБ в ЦНС. Связываясь с активным центром КОМТ он препятствует метилированию катехоламинов (в том числе и дофамина) и скорость метаболизма дофамина замедляется. ФК: Определенным недостатком бромокриптина является его неблагоприятные фармакокинетические свойства. Высокая пресистемная элиминация приводит к тому, что уже во время первого прохождения через печень, после приема внутрь, разрушается 70-80% принятой дозы. Кроме того, период полуэлиминации бромокриптина всего 2-4 часа, что требует его достаточно частого (3 раза) применения в течение суток.
ФК: Определенным недостатком бромокриптина является его неблагоприятные фармакокинетические свойства. Высокая пресистемная элиминация приводит к тому, что уже во время первого прохождения через печень, после приема внутрь, разрушается 70-80% принятой дозы. Кроме того, период полуэлиминации бромокриптина всего 2-4 часа, что требует его достаточно частого (3 раза) применения в течение суток. Перголид (Pergolide, Permax). Механизм действия: Перголид является полным агонистом постсинаптических D2-рецепторов и парциальным агонистом пресинаптических D1-рецепторов. По активности в отношении D2-рецепторов в 10 раз превосходит бромокриптин. За счет активации постсинаптических D2-рецепторов воспроизводит тормозящие эффекты дофамина в экстрапирамидной системе. Действуя на пресинаптические D1-рецепторы, которые расположены на возбуждающих терминалях корковых глутаматергических нейронов, тормозит выделение глютамата и чрезмерную стимуляцию нейронов хвостатого ядра
Перголид (Pergolide, Permax). Механизм действия: Перголид является полным агонистом постсинаптических D2-рецепторов и парциальным агонистом пресинаптических D1-рецепторов. По активности в отношении D2-рецепторов в 10 раз превосходит бромокриптин. За счет активации постсинаптических D2-рецепторов воспроизводит тормозящие эффекты дофамина в экстрапирамидной системе. Действуя на пресинаптические D1-рецепторы, которые расположены на возбуждающих терминалях корковых глутаматергических нейронов, тормозит выделение глютамата и чрезмерную стимуляцию нейронов хвостатого ядра МД: До конца механизм действия амантадина неясен. Полагают, что в реализации его активности играют роль несколько процессов:
МД: До конца механизм действия амантадина неясен. Полагают, что в реализации его активности играют роль несколько процессов: Тригексифенидил (Trihexyphenidyl, Cyclodol Romparkin). МД: Блокирует М- и Н-холинергические рецепторы, которые расположены на тормозных нейронах хвостатого ядра. В итоге, невозможна активация этих рецепторов ацетилхолином, который выделяется возбуждающими холинергическими нейронами хвостатого ядра. Поскольку тормозные нейроны хвостатого ядра остаются неактивными, оно перестает тормозить бледный шар и его активность восстанавливается – он угнетает a-мотонейроны спинного мозга и тонус мышц падает.
Тригексифенидил (Trihexyphenidyl, Cyclodol Romparkin). МД: Блокирует М- и Н-холинергические рецепторы, которые расположены на тормозных нейронах хвостатого ядра. В итоге, невозможна активация этих рецепторов ацетилхолином, который выделяется возбуждающими холинергическими нейронами хвостатого ядра. Поскольку тормозные нейроны хвостатого ядра остаются неактивными, оно перестает тормозить бледный шар и его активность восстанавливается – он угнетает a-мотонейроны спинного мозга и тонус мышц падает. Бипериден (Biperiden, Acineton). По механизму действия, основным фармакологическим эффектам и применению идентичен тригексифенидилу. Отличается несколько более высокой активностью, лучшей способностью устранять тремор и более редкими нежелательными эффектами со стороны ЦНС.
Бипериден (Biperiden, Acineton). По механизму действия, основным фармакологическим эффектам и применению идентичен тригексифенидилу. Отличается несколько более высокой активностью, лучшей способностью устранять тремор и более редкими нежелательными эффектами со стороны ЦНС. Механизм действия: Показано, что фенитоин связывается с инактивированными натриевыми каналами и продлевает это состояние, замедляя реактивацию канала. Таким образом, поступление Na+ внутрь клетки и генерация потенциала действия затрудняется. Поскольку активность нейронов в эпилептогенном очаге значительно выше, чем в нормальной ткани и ПД генерируются там значительно чаще, количество инактивированных натриевых каналов в таких очагах значительно больше, чем в обычных нейронах, поэтому фенитоин подавляет преимущественно активность нейронов эпилептогенного очага.
Механизм действия: Показано, что фенитоин связывается с инактивированными натриевыми каналами и продлевает это состояние, замедляя реактивацию канала. Таким образом, поступление Na+ внутрь клетки и генерация потенциала действия затрудняется. Поскольку активность нейронов в эпилептогенном очаге значительно выше, чем в нормальной ткани и ПД генерируются там значительно чаще, количество инактивированных натриевых каналов в таких очагах значительно больше, чем в обычных нейронах, поэтому фенитоин подавляет преимущественно активность нейронов эпилептогенного очага.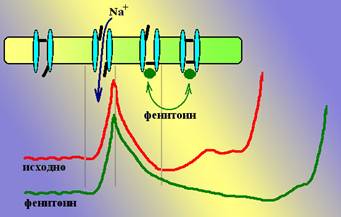 Рисунок 1. Схема влияния фенитоина на натриевые каналы мембраны нейрона. Фенитоин блокирует только инактивированные каналы.
Рисунок 1. Схема влияния фенитоина на натриевые каналы мембраны нейрона. Фенитоин блокирует только инактивированные каналы.
 Карбамазепин (Carbamazepin, Tegretol, Finlepsin). По химическому строению весьма близок к имипрамину. Был введен в клиническую практику в 1960 г, как альтернатива имипрамину, однако, в связи с низкой антидепрессивной активностью он длительно применялся как средство для лечения невралгии тройничного нерва. С 1974 г. используется как противоэпилептическое средство.
Карбамазепин (Carbamazepin, Tegretol, Finlepsin). По химическому строению весьма близок к имипрамину. Был введен в клиническую практику в 1960 г, как альтернатива имипрамину, однако, в связи с низкой антидепрессивной активностью он длительно применялся как средство для лечения невралгии тройничного нерва. С 1974 г. используется как противоэпилептическое средство. Схема 2. Механизм действия карбамазепина и барбитуратов
Схема 2. Механизм действия карбамазепина и барбитуратов
 Этосуксимид (Ethosuximide, Suxilep). Создан в 1960 г. в США. Механизм действия: Этосуксимид блокирует низкопороговые кальциевые каналы (T и N-типа), которые обеспечивают спонтанную деполяризацию мембран таламических нейронов с частотой 3 Гц, выступая в роли своеобразного «водителя ритма». В итоге, нарушается генерация ритмических разрядов таламуса, которая запускает приступ абсанса.
Этосуксимид (Ethosuximide, Suxilep). Создан в 1960 г. в США. Механизм действия: Этосуксимид блокирует низкопороговые кальциевые каналы (T и N-типа), которые обеспечивают спонтанную деполяризацию мембран таламических нейронов с частотой 3 Гц, выступая в роли своеобразного «водителя ритма». В итоге, нарушается генерация ритмических разрядов таламуса, которая запускает приступ абсанса. Фенобарбитал (Phenobarbital, Luminal). Производное барбитуровой кислоты, одно из старейших противоэпилептических средств (для лечения эпилепсии применяется с 1912 г). Длительное время являлся средством выбора при лечении различных форм эпилепсии. В настоящее время его относят к средствам резерва (альтернативным средствам).
Фенобарбитал (Phenobarbital, Luminal). Производное барбитуровой кислоты, одно из старейших противоэпилептических средств (для лечения эпилепсии применяется с 1912 г). Длительное время являлся средством выбора при лечении различных форм эпилепсии. В настоящее время его относят к средствам резерва (альтернативным средствам). Схема 3. Механизм активации ГАМКА-рецептор хлоридного ионофорного комплекса. I – состояние покоя; II – повышение проводимости канала под влиянием ГАМК; III – увеличение частоты открытия канала под влиянием ГАМК на фоне бензодиазепинов (БД-Р – бензодиазепиновый аллостерический центр); IV – увеличение длительности открытия канала под влиянием ГАМК на фоне барбитуратов (Б-Р – барбитуровый аллостерический центр).
По Д.А. Харкевичу, 1999
Схема 3. Механизм активации ГАМКА-рецептор хлоридного ионофорного комплекса. I – состояние покоя; II – повышение проводимости канала под влиянием ГАМК; III – увеличение частоты открытия канала под влиянием ГАМК на фоне бензодиазепинов (БД-Р – бензодиазепиновый аллостерический центр); IV – увеличение длительности открытия канала под влиянием ГАМК на фоне барбитуратов (Б-Р – барбитуровый аллостерический центр).
По Д.А. Харкевичу, 1999