|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
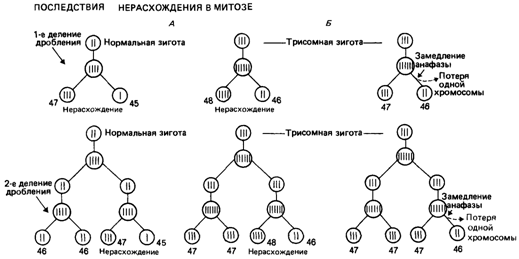
Образование мозаиков по геномным мутациямМозаики по геномным мутациям встречаются довольно часто. Сообщалось, например, что в случае синдрома Дауна один мозаик приходится на 48 пациентов, имеющих стандартную трисомию. Исходя из оценки популяционной частоты синдрома Дауна, равной 1:650, получаем, что частота мозаиков составляет 1:31 000. Она также обнаруживает зависимость от материнского возраста, но в меньшей степени, чем частота простой трисомии по 21-й хромосоме [483]. Механизм образования мозаиков на ранних стадиях дробления. Анализ эффекта материнского возраста позволяет сделать некоторые выводы относительно механизма возникновения мозаиков при синдроме Дауна. Мозаик может развиться из нормальной зиготы. В таких случаях нерасхождение должно происходить в одном из ранних (но не первом) делении дробления 1). Моносомный продукт этого деления обычно теряется. Мозаик может сформироваться также из трисомной зиготы. В этом случае одна клеточная линия должна потерять дополнительную хромосому вследствие анафазного отставания или же должно произойти нерасхождение в соматической клетке (вторичное нерасхождение; рис. 5.28). Можно оценить долю мозаиков, возникших в результате осуществления каждого из этих механизмов. Если они образуются из нормальных зигот, тенденция увеличения возраста матерей не должна проявляться. В случае их возникновения из трисомных зигот следует ожидать увеличения возраста матерей, сходного с тем, которое обнаружено при обобщенном изучении синдрома Дауна. Все мозаики вместе - результат реализации обоих механизмов; средний возраст матерей будет зависеть от доли мозаиков, возникших в результате действия каждой из указанных причин. Проведенный расчет показал, что из 40 мозаиков, описанных в литературе, 20% развились из нормальных зигот. Из этого расчета можно получить сравнительную оценку частоты определенных нарушений митоза, происходящих в нормальных и трисомных зиготах (табл. 5.18). Вычисления показали, что трисомные зиготы обнаруживают почти в 40 раз более сильную тенденцию к анафазному отставанию, чем нормальные клетки, а нерасхождение в первом случае происходит в 70 раз чаще, чем во втором. Однако данные оценки применимы только для мозаиков, развивающихся в индивидов с клинически диагностируемым синдромом Дауна. Вероятность этого намного выше для зигот, бывших первоначально трисомными, чем для зигот, которые при своем образовании были нормальными. На более поздней стадии развития могут возникнуть трисомики с небольшой долей трисомных клеток. Фенотипически они часто бывают нормальными или проявляют лишь слабо выраженные признаки синдрома Дауна, например они могут иметь аномальную дерматоглифику. Они могут быть родителями детей с синдромом Дауна, если участок их яичника или семенника состоит из клеток, имеющих аномальный кариотип. Такие слабовыраженные мозаики, по-видимому, составляют значительную часть родителей детей с синдромом Дауна. 1%-й риск воспроизведения трисомного синдрома Дауна может быть связан с гонадной трисомией по 21 хромосоме того же типа. 1) Нерасхождение в первом делении дробления привело бы к образованию трисомного и моносомного продуктов деления и с потерей моносомной клетки - к возникновению стандартной трисомии 5. Мутации 197
198 5 Мутации 5.1.6.2. Наследственные синдромы с повышенной нестабильностью хромосом [1465; 1464; 1634] Анемия Фанкони (22765). Анемия Фанкони - это детская панмиелопатия, сопряженная с дефицитом костного мозга, приводящим к панцитопении. Больные, как правило, имеют скелетные аномалии, главным образом большого пальца и лучевой кости, и характеризуются гиперпигментацией; часто у них обнаруживают другие пороки развития. Заболевание наследуется по аутосомно-рецессивному типу. Анализ возрастов начала болезни привел к предположению о ее генетической гетерогенности [1638]. Это предположение впоследствии подтвердилось. Было показано, что при слиянии клеток больных с различными клиническими формами патологии происходит взаимная коррекция хромосомной нестабильности [1707]. Существует более распространенная форма, при которой начало болезни приходится на первые годы жизни, и более редкая, когда заболевание возникает в подростковом возрасте. Изучение комплементации между больными, имеющими различные особенности системы репарации [1706] или различное этническое происхождение [1708], не выявило дополнительной генетической гетерогенности. Недавно в ходе исследований клеточной гибридизации были идентифицированы по крайней мере две различные формы этого заболевания. Шредер и др., 1964 [1635] описали двух братьев с этой болезнью - 21 года и 18 лет. Их родители и младший брат (7 лет) были здоровы. У старшего брата обнаружены отклонения от нормального кариотипа: метафазы с множественными хромосомными аберрациями, например с ахроматическими повреждениями (пробелами), хроматидными разрывами; изохроматидными разрывами, ацентрическими фрагментами, дицентрическими хромосомами и хроматидными обменами; в 19 из 39 метафаз выявлено по крайней мере по одной, а в нескольких случаях многочисленные аномалии Эндорепликация отмечена приблизительно в 10% всех метафаз. У среднего брата, не проявляющего клинических симптомов, обнаружено несколько меньшее число митозов с хромосомными аберрациями, однако спектр аномалий такой же, как и у старшего брата Шесть лет спустя и у него развились клинические симптомы заболевания В 32 года больной скончался от множественной геморрагии. При аутопсии у него был обнаружен клинически недиагностируемый рак легких [1634]. Это были первые опубликованные сведения о случаях хромосомной нестабильности при наследственном заболевании. Вскоре данный результат подтвердился при обследовании других больных (рис. 5.29). Синдром Блума (21090). Синдром Блума характеризуется низким весом при рождении, задержкой роста, чувствительностью кожи к солнечному свету и поражением лица телеангиэктазией. Наследуется он по аутосомно-рецессивному типу. Большинство больных родилось в семьях евреевашкенази. Герман и др. [1466], просматривая метафазы в культурах крови семи пациентов, обнаружили у шести из них высокую частоту (4-27%) клеток с разорванными, а иногда и перестроенными хромосомами. При синдроме Блума выявляются и другие цитогенетические аномалии, описанные в случае анемии Фанкони. Отличительный признак синдрома Блума - симметричные четырехлучевые хроматидные обмены, никогда не встречающиеся при анемии Фанкони. По-видимому, они возникли вследствие хроматидных обменов между гомологичными хромосомами. В противоположность этому при анемии Фанкони обычны асимметричные четырехлучевые фигуры, возникшие в результате случайных разрывов негомологичных хромосом. При синдроме Блума частота обменов сестринских хроматид (разд. 2.1.2) была в десять раз выше, чем у здоровых людей или больных с анемией Фанкони. Хотя на первый взгляд эти болезни имеют что-то общее, основные механизмы, приводящие к возникновению синдрома Блума и анемии Фанкони, совершенно различны. Атаксия-телеангиэктазия (20890) [1477]. Двумя постоянными клиническими признаками синдрома атаксии-телеангиэктазии (Луи-Бар) являются прогрессирующая мозжечковая атаксия и глазокожная телеангиэктазия. Атаксия обычно диагнос- 5. Мутации 199
тируется в возрасте 12-14 месяцев; больной оказывается прикованным к инвалидной коляске еще до наступления юношеского возраста. Имеются сообщения о наличии у таких пациентов различных форм иммунодефицита. Самый распространенный дефект иммунной системы – низкий уровень или полное отсутствие IgA. Синдром наследуется по аутосомно-рецессивному типу. Неоднократно публиковались сообщения о хромосомной нестабильности; число разрывов, по-видимому, меньше, чем при анемии Фанкони и при синдроме Блума [1396; 1469; 1485]. Разрывы, вероятно, случайны. Их частота нередко флуктуирует. Анализ хромосом усложняется тем обстоятельством, что фитогемагглютининовая стимуляция лимфоцитов как пра- 200 5. Мутации вило, ослаблена. Часто встречаются псевдодиплоидные клоны; характерной особенностью синдрома является транслокация длинного плеча 14 хромосомы. При всех трех заболеваниях – анемии Фанкони, синдроме Блума и атаксии-телеангиэктазии - увеличение хромосомной нестабильности не артефакт, наблюдаемый in vitro, а феномен, имеющий место in vivo. Резонно предполагать, что клиническая симптоматология этих болезней непосредственно связана с хромосомной нестабильностью. Кроме того, хромосомы больных, страдающих любой из этих трех патологий, проявляют повышенную чувствительность к агентам, разрывающим хромосомы (кластогенным агентам). Хромосомная нестабильность и рак. Пораженные любой из этих трех болезней подвержены сильному риску развития у них злокачественных новообразований. Многие больные с анемией Фанкони в детстве и юности склонны к кровотечениям и инфекциям, имеются сведения и о повышении у них числа неоплазий [1465]. До 1981 г. сообщалось о 45 таких случаях; 22 из них - острая лейкемия (не отмечено ни одной лимфатической формы этого заболевания); 16 - первичные опухоли печени, остальные - карциномы других органов. Все случаи зафиксированы в середине 1960-х гг. после введения стероидной терапии. Не вполне ясно, чем это обусловлено: продлевающим жизнь эффектом такой терапии или тем, что сама стероидная терапия вызывает рак у этих пациентов. Разнообразные злокачественные опухоли были найдены у больных атаксией-телеангиэктазией [1465]. Из 108 пациентов 48 страдали различными «неходжкинскими» лимфомами; 12 - болезнью Ходжкина; 26 - лейкемиями, главным образом лимфатическими, и 22 - другими формами рака (желудка, мозга, яичника, кожи и т. д). Таким образом, преобладают лимфатические неоплазий. У 23 из 99 индивидов с синдромом Блума, о которых было известно до 1981 г., выявлено по крайней мере одно злокачественное новообразование. На основе данных об этих пациентах в молодости было вычислено, что они испытывают в 100 раз больший риск возникновения неоплазий, чем здоровые люди. В отличие от атаксии-телеангиэктазии при этом синдроме наблюдалось громадное разнообразие в распределении опухолей по типам и тканевой локализации. Резонно предположить, что повышенный риск развития неоплазий при этих синдромах может быть прямо связан с повышенной частотой спонтанных хромосомных разрывов. Такая хромосомная нестабильность приводит к появлению большого числа клеток с различными анеуплоидиями, возникшими вследствие разрывов хромосом. Большинство этих клеток гибнет сразу, некоторые претерпевают несколько делений. Однако иногда появляется клетка со структурным дефектом, дающим ей селективное преимущество, частота ее делений более не сдерживается. Такая клетка быстро образует клон генетически одинаковых клеток - первичные раковые клетки. Благодаря своему безудержному росту аномальный клеточный клон будет постепенно замещать нормальные клетки. Если такой клеточный клон содержит хромосому со структурной аномалией, мы должны иногда обнаруживать в определенной части клеток пациентов, страдающих одним из трех синдромов с хромосомной нестабильностью, специфические хромосомные аберрации. Такие клеточные клоны действительно были найдены. На рис. 5.30 приведена фотография необычной хромосомы 1р-, маркирующей клон клеток пациента с анемией Фанкони, наблюдавшего-
5. Мутации 201
ся с 1974 г. [1636]. На рис. 5.31 проведено сравнение частот метафаз с этой маркерной хромосомой по годам. Этот клон, вероятно, имел определенное селективное преимущество, уменьшавшееся, однако, с течением времени. Аналогичные клоны наблюдались и в случае двух других болезней. В случае атаксии-телеангиэктазии наблюдалось полное развитие лейкемии в результате постепенного роста определенного клеточного клона [1485]. Возможные молекулярные механизмы злокачественной трансформации в связи с разрывами хромосом будут обсуждаться' в разд. 5.1.6.5, посвященном онкогенам. Дата добавления: 2015-12-16 | Просмотры: 902 | Нарушение авторских прав |