|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Молекулярные механизмы хромосомной нестабильности и образование опухоли, обусловленное соматической мутациейПигментная ксеродерма (27870-27875). Хромосомная нестабильность и существование маркерных хромосом при трех синдромах, с нею сопряженных, наводят на мысль, что повторный разрыв хромосом может приводить к образованию клеточных клонов, развивающихся в злокачественные опухоли. В связи с этим возникает вопрос о молекулярном механизме хромосомной нестабильности. Много информации на этот счет дало изучение другой наследственной болезни - пигментной ксеродермы. После облучения ультрафиолетовым светом на коже больных с пигментной ксеродермой возникает эритема, которая сменяется атрофией и телеангиэктазией (разд. 3.1.3). Постепенно облученные участки становятся бородавчатыми, и на них развивается рак кожи. Из работ на микроорганизмах известно, что клетки имеют ферментативную систему, способную репарировать дефекты ДНК. Ферментативная репарация дефектов, индуцированных ульт- 202 5. Мутации
рафиолетовым светом, была хорошо изучена на молекулярном уровне в исследованиях на микроорганизмах. Больные пигментной ксеродермой характеризуются аномально высокой чувствительностью к ультрафиолетовому свету. Клэвер [1036, 1420] показал, что причиной этой болезни является нарушение системы репарации ДНК. Позднее было идентифицировано большое число различных ферментативных дефектов, приводящих к фенотипам, подобным тем, что бывают при пигментной ксеродерме (табл. 5.19). Механизмы репарации ДНК. В исследованиях на микроорганизмах изучались три основных механизма репарации ДНК: фотореактивация, эксцизионная репарация и пострепликативная репарация (рис. 5.32) [220; 1458]. 1. Фотореактивация. Сине-фиолетовый свет увеличивает вероятность выживания бактерии, облученной до этого ультрафиолетовым светом. Летальный эффект ультрафиолетового облучения объясняется образованием тиминовых димеров в ДНК, препятствующих ее репликации. Фотореактивирующий фермент эти димеры расщепляет и восстанавливает таким образом правильную структуру ДНК. 2. Эксцизионная репарация. Второй механизм репарации ДНК - эксцизионная репарация - в свете не нуждается. На первом этапе эндонуклеаза опознает димер и разрезает рядом с ним поврежденную цепь ДНК. Образовавшиеся свободные концы распознаются экзонуклеазами, которые расширяют брешь, отщепляя нуклеотиды. Помимо УФ-индуцированных димеров удаляется до 100 других нуклеотидов. Полимераза осуществляет ресинтез удаленного фрагмента цепи, используя в качестве матрицы неповрежденную сестринскую цепь. Наконец, лигаза «сшивает» вновь синтезированный фрагмент со старой цепью. 3. Пострепликативная репарация. Если фотореактивация и эксцизионная репарация по каким-то причинам невозможны, повреждение в цепи не будет исправлено и она не сможет функционировать как матрица в процессе репликации. Во вновь синтезированной комплементарной цепи ДНК останется брешь. Однако генетическая информация, искаженная образованием димеров, содержится в новой цепи ДНК, синтезированной по старой комплементарной цепи. Эта новая цепь и послужит матрицей, на которой образуется копия, замещающая поврежденную цепь ДНК. Точный механизм этого замещения пока неизвестен; по-видимому, он имеет сходство с нормальными рекомбинационными событиями. Одна неповрежденная цепь ДНК необходима в качестве матрицы как для эксцизионной, так и для пострепликативной репарации. Ферментативные дефекты при заболеваниях, сходных с пигментной ксеродермой (27870-27880). Показано, что у культивируемых фибробластов больных пигментной 5. Мутации 203
ксеродермой (ПК) сокращается время жизни после ультрафиолетового облучения. Кроме того, выживаемость различных УФ-облученных вирусов, выращиваемых в ПК-клетках, меньше, чем вирусов, культивируемых в нормальных клетках. Отсюда следует, что в хозяйской клетке существует какой-то генетический дефект, не позволяющий ей исправить дефект вирусного генома. Возможно, у нее нарушен один из вышеупомянутых механизмов репарации. Такое предположение подтвердилось в ходе прямого изучения этих механизмов в ПКклетках. Было обнаружено, что клетки разных больных не способны осуществлять эксцизионную репарацию. У них нарушен начальный ее этап - вырезание димеров. Генетическая гетерогенность [1420; 1421; 1450]. Система эксцизионной репарации включает несколько ферментов, а клинические различия между разными ПК-пациентами свидетельствуют о генетической гетерогенности болезни. Такая гетерогенность обусловлена, по-видимому, тем, что соответствующие мутации могут возникать либо в генах, кодирующих разные полипептидные цепи, либо в различных сайтах одного и того же гена. Один из методов изучения этой проблемы - проведение клеточной гибридизации (разд. 3.4.3) фибробластов от разных больных. Дочерняя клетка, образовавшаяся из двух слившихся клеток, сможет осуществлять эксцизионную репарацию, если ферментативные де- 204 5. Мутации фекты связаны с разными локусами. В этом случае один геном дает один неповрежденный фермент, а другой - второй фермент, т. е. происходит взаимная компенсация двух дефектов. Если ферментативные дефекты идентичны, то даже при повреждении разных сайтов одного гена, компенсация невозможна. С помощью этого метода было идентифицировано по крайней мере восемь групп комплементации (табл. 5.19). Между группами комплементации существуют клинические различия: так, дополнительные неврологические дефекты, например микроцефалию, прогрессирующую умственную отсталость, замедленный рост и половое развитие, глухоту, атаксию, хореоатетоз и арефлексию, имеют только пациенты групп А, В и D (де Санктис и Каккионе, 1932). Многие ПК-пациенты с неврологическими проявлениями не обнаруживают всего спектра симптомов. Даже внутри одной комплементационной группы может существовать удивительная гетерогенность по степени выраженности неврологических симптомов. У многих больных, которым был поставлен клинический диагноз ПК, эксцизионная репарация оказалась совершенно нормальной. Теперь считается, что они страдают определенной формой ПК. У этих пациентов выявлена недостаточность пострепликативной репарации. Дефекты фотореактивации пока не обнаружены. Кроме того, как показано в табл. 5.19, популяционное распределение этих форм очень неравномерно: например, тип А и вариантный тип распространены главным образом в Японии, тогда как тип С, обычный в популяциях европейского происхождения, в Японии редок. Злокачественные новообразования у больных пигментной ксеродермой. У пациентов с ПК рано или поздно развиваются множественные злокачественные опухоли кожи. Облучение УФ-светом может приводить к перерождению клеток любого типа. При этом развиваются базальные и сквамозные клеточные карциномы, злокачественные меланомы, кератоакантомы, гемангиомы и саркомы. Образование опухоли можно предотвратить, либо сведя УФ-облучение к минимуму с помощью специальных средств защиты (например, мазей) либо вообще избегая солнечного света. Повышенный риск возникновения рака у гетерозигот [1340]. Все три синдрома хромосомной нестабильности и пигментная ксеродерма (ПК) наследуются по аутосомно-рецессивному типу. Ферментативная активность у гетерозигот обычно составляет около половины той, что обнаружена у гомозигот по нормальному аллелю (разд. 4.2.2.8). Поэтому имело смысл проверить предположение о возможном увеличении риска возникновения рака у гетерозигот. Наилучшим его подтверждением, которым мы в настоящее время располагаем, могут служить результаты изучения атаксии-телеангиэктазии (А-Т) [1624; 1626]. Это исследование основано на материалах о 27 семьях, объединяющих 1639 близких родственников пробандов с А-Т. Данные о числе случаев рака в этой группе сравнивали с теоретически ожидаемыми для соответствующих контрольных групп (табл. 5.20). Определенное увеличение смертности от рака наблюдается в самой молодой возрастной группе родственников (0 44 года) у представителей разных полов, причем у женщин оно больше, чем у мужчин. Кроме того, у живущих родственников также была обнаружена повышенная частота злокачественных новообразований. При этом выявлено много форм новообразований; как и в случае гомозигот, особенно часто встречались опухоли лимфатической системы, а также карциномы желудка и яичника Согласно полученной оценке, частота гомозигот по атаксии-телеангиэктазии равна приблизительно 1:40000; соответствующая частота гетерозигот составляет ≈ 1%. В этом случае, как показали расчеты, «гетерозиготы по гену А-Т могут включать более 5% всех индивидов, умирающих от рака в возрасте до 45 лет, и около 2% тех больных, которые умирают от него между 45 и 75 годами». Помимо увеличенного риска возникновения рака, гетерозиготы по гену А-Т могут также иметь несколько повышенную предрасположенность к диабетам, тяжелому сколиозу и дефектам нервной трубки [1624]. Другое исследование предрасположенности к раку проводилось на близких родственниках больных с пигментной ксеродермой. Общего увеличения смертности от рака у них не обнаружено. Однако у этих гетерозигот была повышена частота (не приводящих к смерти) немеланомных опухолей кожи [1316]. Это исследование обобщало данные о 2597 близких родственниках ПК-пациентов из 31 семьи, проживающей в США. Представляется довольно интересным, что увеличение частоты случаев заболевания тс- 5. Мутации 205
ми или иными формами рака кожи обнаружено только у жителей южных штатов США, где много солнечного света. Этот факт вместе с отрицательными результатами, полученными для всех других форм рака, кроме карцином кожи, свидетельствует о специфическом дефекте системы репарации УФ-повреждений в эпителиальных клетках, проявляющемся только тогда, когда кожа подвергается сильному облучению солнечным светом, то есть отмеченное увеличение частоты является, так сказать, экогенетическим феноменом (разд. 4.5.2). С другой стороны, в случае анемии Фанкони тщательный анализ имеющихся данных не выявил сколько-нибудь заметного повышения риска возникновения рака у гетерозигот. Молекулярные механизмы синдромов с повышенной хромосомной нестабильностью. Образование тиминовых димеров происходит только в одной из двух сестринских цепей ДНК. Поэтому оно не приводит к немедленному появлению хромосомной бреши или разрыва. Однако если димер не может быть вырезан, цепь, в которой он находится, не будет функционировать в качестве матрицы в ходе следующей репликации, комплементарная ей цепь ДНК окажется неполной и во втором цикле репликации появится видимый разрыв (рис. 5.33). Следовательно, если брешь в двойной цепи ДНК связана с видимыми в микроскоп хромосомными разрывами, следует ожидать большего увеличения числа хромосомных разрывов после облучения ПК-клеток, чем нормальных клеток. Такое увеличение действительно было описано. С другой стороны, в необлученных ПК-клетках нестабильности хромосом не наблюдалось. Этим они отличаются от клеток больных с анемией Фанкони, синдромом Блума и атаксией-телеангиэктазией. Эти заболевания всегда сопровождаются спонтанной хромосомной нестабильностью. Следовательно, молекулярные дефекты, лежащие в их основе, различны. Разумно, однако, предполагать, что определенные нарушения механизмов репликации и репарации ДНК также могут быть в числе факторов, приводящих к возникновению этих синдромов. Некоторые данные подтверждают сделанный вывод. Однако, несмотря на все предприни-
206 5. Мутации
мавшиеся усилия, глубоко разобраться в молекулярных основах этих синдромов до сих пор не удалось. Для их объяснения предложено много разных механизмов. Их связывают с дефектами различных ферментов репарации, пониженным содержанием кофакторов ферментов, аномальным транспортом ферментов, необходимых для репликации ДНК (например, топоизомеразы) через ядерную мембрану или недостаточными энергетическими запасами. Были установлены некоторые интересные факты, по-видимому, важные в функционировании такого механизма, например замедление роста цепи ДНК при синдроме Блума. Однако главные, определяющие дефекты пока неизвестны. Их выявление имело бы важное значение, так как эти синдромы служат моделями как при выяснении молекулярных механизмов спонтанных мутаций вообще, так и молекулярных механизмов соматических мутаций, связанных с новообразованиями, в частности. Цепь событий при образовании злокачественных неоплазий, возникающих в результате соматической мутации [1633]. Цепочка событий, приводящих к образованию неоплазий, обусловленных соматической мутацией, изображена на рис. 5.34. Первый этап - повреждение ДНК. Оно может быть вызвано или внутренними факторами, например нарушением механизмов репликации или репарации, или внешними, например ионизирующей радиацией, химическими мутагенами или вирусами. Повреждение ДНК способно полностью вывести из строя репликацию и поэтому может быть летальным. Другая возможность заключается в том, что повреждение будет репарировано, но с ошибкой. Здесь не имеет принципиального значения, какого типа мутации при этом образуются. На-
5 Мутации 207
пример, это может быть точковая мутация, возникшая в результате замены одного основания, или видимая хромосомная аномалия. Мутация может оказаться летальной и привести к гибели несущий ее клеточный клон. Другая возможность заключается в том, что клетки, содержащие мутацию, будут расти нормально; в этом случае единственным индикатором мутаций окажется маркерная хромосома. Наконец, не исключена вероятность того, что новый клеточный клон будет обладать селективным преимуществом, обусловленным генетическими дефектами нормальных механизмов торможения и регуляции роста. В этом случае развивается рак. Причиной возникновения опухоли могут быть и вторичные генетические процессы, например образование дополнительных анеуплоидий. В некоторых случаях они вызывают гибель клеток, а иногда приводят к появлению клонов, имеющих селективное преимущество. Неконтролируемый рост новообразования продолжается до тех пор, пока больной не погибнет от нарушения нормальной жизнедеятельности. Мысль о том, что хромосомы могут играть какую-то роль в возникновении опухолей, высказывалась еще в начале нашего века Бовери (1914, см. [77]). Доказать эту гипотезу удалось лишь после появления тонких цитогенетических методов. 5.1.6.4. Другие факты, свидетельствующие о роли соматической мутации в механизме канцерогенеза [1520] История мутационно-соматической гипотезы возникновения рака. Наследственные синдромы с повышенной хромосомной нестабильностью и дефектами репликации и репарации ДНК можно рассматривать в качестве модели для изучения молекулярных механизмов соматической мутации и образования опухоли. Гипотеза о том, что рак может быть обусловлен соматической мутацией, появилась гораздо раньше работ, посвященных изучению этой проблемы. Еще фон Ханземан (1890) [1475] на основании результатов собственных исследований митоза постулировал, что клетка злокачественной опухоли представляет собой клетку с аномальным содержанием хроматина [77]. Бовери (1914) конкретизировал эту идею, выдвинув предположение о неравном распределении хромосом определенной клетки между ее потомками. Однако он подчеркнул, что здесь существенна аномальная хроматиновая конституция как таковая, а не механизм ее возникновения. В течение последующих десятилетий гипотеза соматических мутаций разрабатывалась многими авторами и обсуждалась в различных аспектах. Наиболее четкие формулировки самых важных следствий из нее дал Бёрнет (1957, 1974) [1408]: 1) новообразования должны быть моноклональными, то есть они должны происходить от одной единственной клетки; 2) их частота может повышаться при действии химических агентов или вирусов, взаимодействующих с ДНК; 3) в отдельных клетках из большой популяции пролиферирующих раковых клеток могут возникнуть дополнительные мутации, обусловливающие дополнительные селективные преимущества; субклоны, ведущие начало от этих клеток, будут быстро перерастать скопления других опухолевых клеток; 4) мутационная гипотеза объясняет также увеличение частоты большинства форм рака с возрастом, если возникновение соматических мутаций можно в качестве первого приближения считать процессом, зависящим от времени. Кроме того, клиническому проявлению клонов должно предшествовать несколько лет их роста. Мы не будем больше рассматривать здесь индукцию рака химическими веществами, реагирующими с ДНК. Достаточно отметить, что многие мутагены (раздел 5.2.2) оказались и сильными канцерогенами. Злокачественные неоплазии обычно состоят из субклонов, имеющих разные кариотипы, что свидетельствует о множественных аномалиях митозов во время пролиферации опухоли. Увеличение частоты новообразований с возрастом - хорошо известная общая закономерность биологии рака. В настоящее время мы располагаем 208 5. Мутации
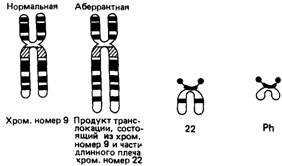
многочисленными данными, доказывающими моноклональное происхождение опухолей. Известно, например, что один В-лимфоцит продуцирует легкие цепи углобулина только одного специфического типа - λ или ϰ. Однако разные В-лимфоциты синтезируют легкие цепи, отличающиеся по «вариабельной» части аминокислотной последовательности (разд. 4.4). С другой стороны, при миеломатозе - одном из раковых заболеваний - все эти клетки продуцируют легкие цепи с идентичными вариабельными участками. У женщин, гетерозиготных по двум вариантам гена G6PD, сцепленного с Х-хромосомой (разд. 2.2.3.3), мышечная ткань матки является мозаичной: разные клетки экспрессируют разные варианты фермента, как это и ожидалось на основе предположения о случайной инактивации Х-хромосом. С другой стороны, фиброидные опухоли матки всегда содержат один и тот же вариант во всех клетках. Аналогичные факты известны также и для других опухолей [1519]. Большинство опухолей действительно имеют моноклональное происхождение. Некоторые наследственные опухоли, например нейрофибромы, имеют мультиклональное происхождение, из чего следует, что тенденция к пролиферации свойственна каждой клетке [1449]. Вирусная этиология или соматическая мутация? В настоящее время существует много наблюдений, сделанных главным образом на животных, из которых явствует, что причиной возникновения опухолей могут быть вирусы. Резонно предполагать, что некоторые опухоли человека также имеют вирусное происхождение. Эта гипотеза не противоречит гипотезе соматических мутаций. Вирусы часто бывают сайт-специфическими и могут индуцировать в хромосоме то или иное мутационное событие. Поэтому процесс образования опухоли, следующий за вирусным повреждением, возможно сходен с аналогичным процессом, описанным в случае соматических мутаций любого типа. Неоплазии, сопряженные с определенными хромосомными аберрациями. Существование единственной специфической хромосомной аберрации, имеющейся только в опухолевой ткани, было описано еще много лет назад для некоторых неоплазии. Классическим примером такой аберрации является филадельфийская (Ph1) хромосома, которая почти всегда наличествует у больных хроническим миелоидным лейкозом [1584]. При этом заболевании обычно обнаруживают транслокацию почти всего длинного плеча 22-й хромосомы на 9-ю [1478] (рис. 5.35). Такие пациенты имеют нормальные хромосомы во всех тканях, за исключением системы гематопоэза. Фактически же специфическая аномалия хромосом оказывает влияние на все клетки - предшественницы клеток крови, включая мегакариоцитные и эритропоэтические клетки. К аналогичным выводам привело исследование, в котором в качестве биохимического маркера использовали варианты G6PD. Однако клинические и гематологические последствия этой транслокации проявляются только в гранулоцитарных элементах крови; это свидетельствует о том, что данная «мутация», несмотря на ее присутствие в клетках нескольких типов, может влиять на характер роста лишь одной дифференцированной ткани. В некоторых, составляющих исключение семьях от хронического миелоидного лейкоза умирало по нескольку человек, а в одной семье [1488] несколько ее младших представителей имело в своих гематопоэтических клетках Ph1-хромосому, не проявляя клинических признаков лейкоза. В этой семье склонность к разрыву 22-й хромосомы наследуется по аутосомнорецессивному типу. Менингиомы – это опухоли одной из трех оболочек мозга. Они гистологически доброкачественны и в большинстве случаев состоят из гиподиплоидных клеточных линий, в которых отсутствует или имеет делецию одна из хромосом 22. На последующих стадиях образования субклонов возникают изменения других хромосом [1709-1711; 1545]. Разработка нового, высокоразрешающего метода дифференциального окрашивания хромосом (разд. 2.1.2.3), усовершенствование методов приготовления хромо- 5. Мутации 209
сомных препаратов из отдельных клеток твердых опухолей позволили обнаруживать специфические хромосомные аберрации во все большем и большем числе других неоплазий [1704]; в большинстве случаев были идентифицированы или делеция полосы (субполосы) или реципрокная транслокация между двумя хромосомами. В табл. 5.21 перечисляются опухоли, для которых идентифицированы специфические транслокации и точки разрыва. Вполне возможно, что аберрации имеются и в тех случаях, когда их не нашли. Просто они могут быть так малы, что не будут выявляться даже с помощью дифференциального окрашивания. Почему же, однако, к селективному преимуществу клетки приводят лишь определенные хромосомные аберрации и почему это преимущество является тканеспецифическим? В последние годы молекулярным биологам удалось найти частичный ответ на первый из вопросов (разд. 5.1.6.6). Но, прежде чем рассказывать об этих результатах, мы рассмотрим один пример, касающийся солидной опухоли. Ретинобластома (18020). Ретинобластома представляет собой детское офтальмологическое раковое заболевание. Ретинобластома может иметь наследственную и ненаследственную природу [1669; 1676]. 210 5. Мутации
Наследственная форма является аутосомно-доминантной с приблизительно 90%-ной пенетрантностью; в разных семьях пенетрантность варьирует [1548]. Приблизительно 68% всех случаев наследственной формы заболевания носят билатеральный характер, остальные - унилатеральный. В некоторых билатеральных случаях во втором глазу обнаружено более одной первичной опухоли [1519-1522]; в унилатеральных случаях, как наследственных, так и ненаследственных, множественное возникновение опухолей обычно не наблюдалось. Многие пациенты, страдающие ретинобластомой, являются спорадическими больными, т.е. они первые и единственные пораженные в своих семьях. У этой категории больных билатеральное поражение встречается только в ~ 20-25% случаев Спорадические больные принадлежат к одной из двух групп: первая включает тех, болезнь которых обусловлена доминантными мутациями de novo и 50% потомков которых страдает той же патологией, а вторая - больных ненаследственной формой заболевания. Все спорадические больные с билатеральным поражением появились в результате мутаций de novo; сегрегационное отношение для их потомков не 5. Мутации 211
намного ниже 50% [1628]. Из спорадических случаев с унилатеральным поражением около 10-12% возникли вследствие мутаций de novo, а остальные - больные ненаследственной формой. В одной из работ описан ряд следствий для генетического консультирования, вытекающих из этой ситуации [1676] (см., например, приложение 8). Две мутационные стадии при наследственной форме заболевания. Предполагается, что становление наследственной формы болезни происходит в два этапа. На первом этапе мутирует половая клетка. Эта мутация приводит к таким изменениям хромосомной структуры, которые увеличивают риск возникновения соматической мутации. Мутация в соматической клетке трансформирует одну или несколько измененных клеток в клетки ретинобластомы. Для возникновения ненаследственной ретинобластомы необходимы, как принято считать, две соматические мутации: на первой стадии клетка может превратиться в потенциальную клетку ретинобластомы; в результате ее состояние может теперь стать идентичным состоянию любой клетки наследственной ретинобластомы. Это означает, что опухоль развивается только в том случае, если за первой стадией следует вторая, возможно эквивалентная одному из этапов, необходимых для становления наследственной формы заболевания. Еще в 1963 г была описана делеция 13q. Ее обнаружили во всех клетках пациента с билатеральной ретинобластомой и не очень значительными конституциональными аномалиями [1531]. В последние годы, особенно после введения дифференциального окрашивания высокого разрешения, выявлено большое число таких пациентов; у многих из них делетирован или вовлечен в реципрокную транслокацию лишь очень небольшой участок длинного плеча 13-й хромосомы (рис. 5.36). Сравнивая множество подобных фактов, можно установить, что делетированный сегмент приурочен к 13q14 (или даже к 13q14.13; рис. 5.36). «Ген» ретинобластомы (если мы назовем данный участок хромосомы геном) тесно сцеплен с геном эстеразы D (ESD). В ходе исследований возник другой вопрос: может ли вторая стадия, обусловливающая развитие злокачественного клеточного клона, заключаться в делении хромосомного сегмента в районе 13q14. В этом случае такая делеция должна выявляться во всех опухолевых клетках. По методическим причинам, изучение хромосом опухолевых клеток долгое время представляло определенные трудности; кроме того, клетки солидных опухолей основательно перемешаны с клетками соединительной ткани. Именно этим можно объяснить тот факт, что данные одной старой работы, в которой сообщалось об изменениях сегмента 13q в опухолевых клетках, вскоре после ее появления были опровергнуты. Впоследствии однако вывод о важной роли участка
212 5. Мутации
13q14 подтвердился в ходе изучения многих клеток пациентов с наследственной и ненаследственной формами заболевания [1380]. Даже если бы этот вывод оказался справедливым для большинства случаев, это не решило бы проблему проявления мутации лишь в немногих клетках из тех, что ее содержат. Какую-то ясность в этот вопрос могут внести молекулярно-биологические исследования (разд. 2.3) [1389; 1414; 1476] с использованием полиморфизма рестрикционных фрагментов ДНК, тесно сцепленных с геном ретинобластомы. Согласно полученным данным, решающая стадия протекает в гомологичной хромосоме, несущей нормальный аллель. Иногда рестрикционные маркеры, унаследованные от одного из родителей, в раковых клетках полностью отсутствовали, что свидетельствует об исчезновении хромосомы, полученной от этого родителя, и ее замещении второй копией хромосомы, несущей мутантный ген ретинобластомы; в других случаях к тому же следствию - переводу мутации в гомозиготное состояние - приводят рекомбинационные события, в которых принимает участие только часть нормальной хромосомы (рис. 5.37, 5.38). Значение этого результата, по-видимому, выходит за рамки частного случая, при изучении которого он был получен. Такие события, как нерасхождение или рекомбинация, возможно, происходят в соматической ткани гораздо чаще, чем до сих пор предполагалось. С лх помощью можно объяснить различные особенности фенотипов и даже механизм возникновения болезней. В частности, его можно использовать для объяснения других аутосомнодоминантных опухолевых заболеваний. Генетические синдромы, сопряженные с опухолями. У пациентов с ретинобластомой часто возникает остеосаркома. У таких больных, как и у всех пациентов с ретинобластомой, обнаружена гомозиготность по тому же локусу 13-й хромосомы [1525]. Таким образом, гомозиготность по одному и тому же сайту может быть причиной как ретинобластомы, так и остеосаркомы. В опухоли Вилмса, как и предсказывал Кнудсон, сходные механизмы привели к гомозиготизации одного сайта на 11-й хромосоме, а именно 11р13. У этих больных обнаружен редкий аутосомно-доминантный синдром, характеризующийся аниридией, мочеполовыми аномалиями (гонадобластомой, недифференцированными гениталиями) и умственной отсталостью. Тот же локус, по-видимому, участвует в форми-
5 Мутации 213
ровании синдрома Беквита - Видемана, который сопряжен с различными эмбриональными опухолями (табл. 5.22). Общий механизм возникновения этих разных опухолей также включает соматическую гомозиготизацию в сайте 11-й хромосомы [1525]. Предполагается, что этот аллельный сайт наследуется от одного из родителей. Опухолевые заболевания бывают как ненаследственными с возникновением единичных опухолей, так и наследуемыми по доминантному типу с образованием множественных опухолей. Хорошо известно, что такая ситуация имеет место в случае опухолей кожи [1631]: одиночные опухоли обычно ненаследственны, тогда как множественные опухоли того же гистологического типа - доминантно наследуемый признак. В качестве примеров такого рода можно привести нейрофиброматоз и одиночные нейрофибромы, множественные и одиночные липомы, множественные и спорадические кожные лейомиомы, множественные и спорадические клубочковые опухоли, а также синдром базальноклеточного невуса и одиночные базальноклеточные невусы [1391]. Кнудсон представил аналогичные эпидемиологические и статистические данные, свидетельствующие о том, что сходные механизмы могут действовать при образовании многих других опухолей у детей, например нейробластомы и феохромоцитомы. Постулируется существование как спорадических, так и наследственных случаев этих заболеваний. Наследственные случаи чаще носят семейный характер, часто бывают билатеральными, приурочены к более раннему возрасту и характеризуются тем, что опухоль возникает более чем в одном месте (так, например, при нейробластоме – в обоих надпочечниках) [1519; 1522; 1549]. В будущем можно будет локализовать гены, ответственные за возникновение различных сопряженных с опухолями генетических синдромов, путем идентификации хромосомных сайтов, в которых в процессе онкогенеза происходит специфическая по- 214 5. Мутации
теря конституциональной гетерозиготности. Несколько примеров таких генетических синдромов с указанием сопряженных с ними опухолей дается в табл. 5 22. «Раковые семьи» известны и в случае других злокачественных опухолей. Данные о карциноме пищевода в семьях, характеризующихся специфическим типом кератодермии ладоней и подошв, обсуждаются в разд. 3.6 (рис. 3.68). Этот пример, как и другие случаи доминантного наследования предрасположенности к возникновению неоплазий, показывает, что в раковых семьях проявляется тенденция к более раннему формированию опухолей в жизни больных, чем при более распространенной спорадически возникающей разновидности того же ракового заболевания. Часто обнаруживается выраженная тенденция к множественному образованию опухолей у одного индивида. Кроме того, эти факты свидетельствуют о генетически детерминированной и доминантно наследуемой повышенной вероятности злокачественной трансформации. 5.1.6.5. Онкогены [1686; 1690, 1691, 1696} Основные принципы. Выполненные в последние годы молекулярно-биологические исследования и особенно открытие так называемых онкогенов способствовали пониманию молекулярных механизмов канцерогенеза. В ранних работах по слиянию клеток (разд. 3.4.3) было показано, что важную роль в злокачественной трансформации могут играть мутации в определенных локусах и обработка клеток хомячка ДНК вируса полиомы С другой стороны, со времени появления работ Эллермана и Бэнга (1908) и Рауса (1911) стало известно о способности некоторых РНК-вирусов - ретровирусов - индуцировать опухоли у животных. Эти и полученные позднее результаты побудили многих исследователей заняться поиском ретровирусов, которые могли бы вызывать опухоли у человека. До конца 1970-х г. эти поиски были безуспешными. Однако внедрение в практику исследований молекулярно-биологических методов позволило получить в последние годы очень важные результаты. Показано, что геном ретровируса состоит из одноцепочечной РНК. Он включает следующие информационные последовательности (от 5'- к 3' концу): 5'-регуляторную последовательность; гены, кодирующие белки, необходимые для формирования внутренней структуры; ген обратной транскриптазы; гены поверхностных гликопротеинов; З'-регуляторную последовательность. Как только вирусная частица проникает в клетку, обратная транскриптаза осуществляет синтез двухцепочечной ДНК-копии одноцепочечной РНК. Затем ДНК-копия встраивается в хромосомную ДНК клетки; интеграция может происходить во многих сайтах генома хозяина. Эта ДНК вызывает в клетке синтез новой вирусной РНК и белков, необходимых для синтеза новых вирусных частиц. Помимо этой минимальной информации геномы онкогенных ретровирусов несут дополнительный ген, являющийся специфическим фактором, вызывающим злокачественную трансформацию хозяйских клеток. Этот ген называется ретровирусным онкогеном (v-onc). В исследованиях по 5. Мутации 215
гибридизации (разд. 2.3) с использованием ДНК-зондов на основе генов v-onc было показано, что такие гены гомологичны генам, находящимся в различных сайтах хозяйского генома. Однако при нормальных условиях они не приводят к злокачественной трансформации. Эти гены называются протоонкогенами или клеточными онкогенами (с-опс). В недавно опубликованном обзоре [1696] упомянуто несколько генов с-опс, которые расположены в различных аутосомах человека (табл. 5.23; см. также табл. П9.5). В настоящее время полагают, что эти гены были интегрированы вирусным геномом на каком-то этапе эволюционного процесса. Однако совершенно неизвестно, почему они вызывают злокачественную трансформацию при переносе в клетку в результате вирусной инфекции и не вызывают таковой, когда передаются как нормальные клеточные компоненты. Три разных гена с-опс расположены в 3, 15 и 20-й хромосомах человека и кодируют три тирозинспецифические протеинкиназы, давая гомологичные по аминокислотным последовательностям генные продукты. Эти протеинкиназы фосфорилируют белки и таким образом влияют на их биологическую активность, что в конечном итоге
приводит к трансформации в результате, например, изменения свойств клеточной поверхности (контактного торможения?). Протоонкогены могут кодировать нормальные факторы роста или их рецепторы. Так, ген sis кодирует одну цепь матрицы, на которой синтезируется фактор роста, а erb-b - матрицу для синтеза рецептора эпидермального фактора роста [1437]. Таким образом, протоонкогены способны через различные факторы роста и их рецепторы контролировать нормальный рост клеток. Можно легко выяснить, как именно мутантные протоонкогены (см. ниже) стимулируют митоз и вызывают рост раковых опухолей. Особый интерес представляет фактор роста, синтезируемый на матрице [1437]. По-видимому, он участвует в образовании атероматозных очагов: предполагается, что они формируются в результате повышенного синтеза нормального фактора роста. Его мутантный аллель sis участвует в образовании неопластической саркомы. Обнаружение связи между генами, кодирующими нормальные факторы роста, и их мутантными аллелями, т. е. онкогенами, индуцирующими опухоли, имеет большое значение для биологии развития и биологии рака [1694]. Существуют структурные различия между v-onc и гомологичными им генами с-опс; так, например, гены с-опс состоят, подобно другим эукариотическим генам, из экзонов и интронов (разд. 2.3), тогда как соответствующие гены v-onc сохранили только экзоны. Клеточная трансформация. Во многих случаях клеточные онкогены были выявлены путем прямого переноса от трансформированных клеток к нормальным. Обработка пренеопластических фибробластов мышей NIH/3T3 ДНК из опухолегенных клеточных линий в определенной части случаев приводит к появлению трансформированных опухолевых фенотипов в реципиентных клетках. Первый трансформирующий ген, охарактеризованный в трансформантах NIH/3T3 (c-Ha-ras1), был найден в клеточной линии EJ карциномы мочевого пузыря человека. Генетическое повреждение, приводящее к активации, т. е. к появлению трансформирующей способности 216 5 Мутации
онкогена, представляет собой, как было установлено с помощью клонирования, единичную точковую мутацию, обусловливающую замещение одной аминокислоты. Кодон GGC замещается кодоном GTC; трансверсия G ® Т приводит при возникновении мутантного белка к замене глицина валином. Однако поиск этого протоонкогена у больных 29 формами рака в ходе анализа с применением ферментов рестрикции не привел к обнаружению случаев носительства; вероятно, данный протоонкоген редок и сопряжен лишь с немногими раковыми болезнями. Интересно, что у вирусной копии этого онкогена из вируса мышиной саркомы выявляется точковая мутация, возникшая в той же самой позиции. Были обнаружены другие случаи активации таких генов в результате единичной точковой мутации. Кроме того, некоторые онкогены, найденные в других опухолях, проявляли структурное сходство с геном c-Ha-ras1. Возможно, что мутантные аллели генов ras причастны к возникновению ≈ 15% опухолей человека. Из клеточных линий лимфомы Беркитта был выделен трансформирующий ген, имеющий иную структуру (В-lym); продукт этого гена частично гомологичен трансферрину - белку, переносящему железо (см. разд. 6.1.2). В клеточных линиях лимфомы Беркитта обнаружена также перестройка другого гена - с-тус. Тот факт, что в опухоли одного типа найдено два различных онкогена, свидетельствует о том, что иногда для злокачественной трансформации, может быть необходимо возникновение мутаций более чем в одном локусе. Разные стадии канцерогенеза, вероятно, отражают последовательную активацию различных протоонкогенов, приводящую к качественным и количественным изменениям экспрессии генов. В настоящее время идет очень интенсивное изучение механизмов такой активации, поскольку их выяснение обещает прогресс в понимании молекулярных процессов, приводящих к злокачественной трансформации. Согласно исследованиям in vitro, кроме вышеупомянутых точковых мутаций, трансформирующий эффект оказывает присоединение генов с-опс к участкам ДНК, являющимся сильными промоторами или энхансерами, например в результате инсерции таких участков по соседству с генами с-onc. Однако вставка промотора (или энхансера) рядом с онкогеном, возможно, является только одним из необходимых условий трансформации, которая завершается в результате изменений в других протоонкогенах. Участвуют ли онкогены в канцерогенезе, обусловленном хромосомными перестройками? При рассмотрении результатов изучения онкогенов (особенно данных о том, что инсерция онкогенов рядом с сильными промоторами приводит к их активации) возникает вопрос: может быть при хромосомных перестройках, специфичных для определенных неоплазий (раздел 5.1.6.4), решающую роль играют перемещения онкогенов в области, соседние с промоторно/энхансерными участками (и, возможно, в области, примыкающие к другим регуляторным генам)? Поэтому в настоящее время многие группы исследователей занимаются изучением онкогенов и их активности в опухолях при локализации в нормальных и перестроенных хромосомах (рис. 5.39). При лимфоме Беркитта, например, может происходить 20-кратное увеличение транскрипции гена с-тус [1704]. В плазмацитомах мышей обнаружена транслокация, сходная с той, которая у людей приводит к лимфоме Беркитта, между терминальным участком хромосомы 15, несущим с-тус, и хромосомой 12. Точка разрыва совпадает с константным участком гена тяжелой цепи иммуноглобулина. Сходная ситуация, по-видимому, имеет место в случае лимфомы Беркитта у человека. Онкоген с-abl человека локализуется в терминальном диске длинного плеча хромосомы 9 - в том самом диске, который расположен в точке разрыва, происходящего при транслокации 9:22 (она сопряжена с хроническим миелоидным лейкозом). Пока слишком рано делать окончательные выводы; однако предварительные данные свидетельствуют в пользу гипотезы о том, что активация онкогенов действительно может играть определенную роль в канцерогенезе, обусловленном хромосомными перестройками Протоонкогены обнаружены также в виде 5. Мутации 217
амплифицированных копий в опухолевых клетках [1694]. Цитологическим проявлением генной амплификации, имеющей место в раковых клетках, являются удвоенные мелкие хромосомы и одинаково окрашивающиеся участки. Онкоген myc, транслоцируемый при лимфоме Беркитта, обнаруживает повышенную экспрессию в случае карциномы легких, карциномы кишечника и промиелоидного лейкоза. Ген N-myc амплифицируется на поздних стадиях развития нейробластомы; высокие уровни фактора роста эпидермиса при сквамозноклеточных карциномах, по-видимому, объясняются амплификацией гена erb-b. Опухолевый рост может быть связан с активностью специфических онкогенов, поскольку в интенсивно растущих формах опухолей обнаруживаются определенные онкогены. Аналоги онкогенов или антионкогенные антитела в противоположность современной химиотерапии рака могут приостановить опухолевый рост без нарушения функционирования нормальных клеток. Изучение онкогенов - по крайней мере тех, которые известны в настоящее вре- 218 5. Мутации мя, - конечно, не дает полного ответа на вопрос о механизмах канцерогенеза. Так, в большинстве опухолей человека активированные онкогены не найдены (может быть, потому, что не все онкогены еще известны). К тому же пока совершенно не решен вопрос о том, почему инфицирование ретровирусами, содержащими онкогены, как правило, приводит к злокачественному перерождению только одной определенной ткани. Такие гены должны проявлять строго тканеспецифическую экспрессию. Тем не менее открытие онкогенов было важным шагом на пути к пониманию природы злокачественной трансформации. Дата добавления: 2015-12-16 | Просмотры: 1574 | Нарушение авторских прав |










 ); стрелками справа от хромосом отмечены специфические диски, подвергающиеся транслокациям (
); стрелками справа от хромосом отмечены специфические диски, подвергающиеся транслокациям ( ) или делециям (
) или делециям ( ), обнаруживаемым у пациентов, страдающих перечисленными заболеваниями. Дополнительные обозначения (
), обнаруживаемым у пациентов, страдающих перечисленными заболеваниями. Дополнительные обозначения ( ) указывают на связь той или иной аберрации с определенной болезнью. ЛБ-лимфома Беркитта; К А - карцинома; ОЛЛ - острая лимфобластическая лейкемия; ОНЛЛ - острый нелимфоцитарный лейкоз; ОМЛ- острый миелоидный лейкоз; ХМЛ- хронический миелоидный лейкоз; ОММЛ- острый миеломоноцитарный лейкоз; ОМОЛ -острый монобластический лейкоз; БК – бластический криз; НХЛ - неходжкинская лимфома; ХЛЛ - хронический лимфолейкоз; ОПЛ – острая промиелоцитарная лейкемия; МПР – миелопролиферативное расстройство. Буквой Р обозначено короткое плечо, a q- длинное плечо хромосомы; t, del и inv - соответственно транслокация, делеция и инверсия [1705].
) указывают на связь той или иной аберрации с определенной болезнью. ЛБ-лимфома Беркитта; К А - карцинома; ОЛЛ - острая лимфобластическая лейкемия; ОНЛЛ - острый нелимфоцитарный лейкоз; ОМЛ- острый миелоидный лейкоз; ХМЛ- хронический миелоидный лейкоз; ОММЛ- острый миеломоноцитарный лейкоз; ОМОЛ -острый монобластический лейкоз; БК – бластический криз; НХЛ - неходжкинская лимфома; ХЛЛ - хронический лимфолейкоз; ОПЛ – острая промиелоцитарная лейкемия; МПР – миелопролиферативное расстройство. Буквой Р обозначено короткое плечо, a q- длинное плечо хромосомы; t, del и inv - соответственно транслокация, делеция и инверсия [1705].