|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
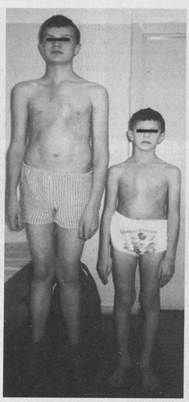
Синдром Марфана и другие наследственные синдромы с марфаноподобным фенотипомСиндром Марфана впервые описан в 1896 году французским педиатром А. Б. Марфаном. У больных наблюдается одновременное поражение трех систем: опорно-двигательной, сердечно-сосудистой и органа зрения. Характерными клиническими проявлениями синдрома Марфана являются высокий рост, арахнодактилия (длинные, тонкие, «паукообразные» пальцы рук), гиперподвижность суставов, подвывих хрусталика и миопия, поражение крупных сосудов (аневризма аорты), порок сердца (пролапс митрального клапана. Каждый из этих симптомов может отличаться по степени тяжести и сочетаемости друг с другом у отдельных членов семьи.
Рисунок 3. Родные братья с синдромом Марфана.
Для синдрома Марфана характерны выраженная плейотропия, варьирующая экспрессивность и высокая пенетрантность. Диагноз синдрома Марфана, ставится при наличии минимум пяти симптомов - аневризма аорты, вывих хрусталика, арахнодактилия, деформация грудины, кифосколиоз. При этом имеет место увеличение (в два раза и более) выведения с мочой глюкозоаминогликанов и их фракций. Особенно резко возрастает почечная экскреция хондроитин-4-6-сульфатов и в меньшей степени - гиалуроновой кислоты и гепаран-сульфата. В моче больных определяется также повышенное содержание (в два и более раз) аминокислоты оксипролина. Распространенность заболевания в разных популяциях варьирует в пределах от 1:5 до 1:25 тысяч населения. Причиной развития классических форм заболевания 1 типа являются гетерозиготные мутации в гене фибриллина 1 - корового гликопротеина микрофибрилл эластических волокон внеклеточного матрикса, выполняющего в большинстве соединительных тканей архитектурные функции. Ген фибриллина 1 (FBN1) картирован в области 15q21.1, и в настоящее время в нем идентифицировано более 550 мутаций. Эти мутации обладают широким спектром клинических проявлений от изолированной эктопии хрусталика с мягкими скелетными проявлениями марфаноподобного типа до тяжелых неонатальных форм синдрома Марфана, заканчивающихся летальным исходом в течение первых двух лет жизни. При этом мутации, ассоциированные с тяжелыми формами синдрома, локализованы в определенных экзонах гена FBN1. Молекулярно-генетическая диагностика синдрома Марфана как в пренатальном, так и в постнатальном периоде принципиально возможна, но осложняется тем обстоятельством, что мутации в гене фибриллина уникальны, то есть описаны у больных только одной или значительно реже двух или нескольких неродственных семей. Подавляющее большинство из них приводят к классическим формам синдрома Марфана. Однако описаны другие аллельные варианты синдрома, выделяемые клиницистами в самостоятельные нозологические формы -табл. 1. Важную роль в патогенезе синдрома Марфана играет трансформирующий фактор роста бета (Tgf|3), латентная форма которого непосредственно связана с фибриллином 1. Предполагается, что при снижении уровня фибриллина 1 повышается активность Tgf|3, следствием чего является высвобождение протеаз, участвующих в медленной деградации эластических волокон и других компонентов внеклеточного матрикса. Однако эти предположения требуют дополнительного экспериментального подтверждения. Наряду с синдромом Марфана и его аллельными вариантами, известны другие марфаноподобные аутосомно-доминантные заболевания, не связанные с мутациями в гене FBN1. Так, мутации в гене FBN2, кодирующем изоформу 2 фибриллина, найдены у больных синдромом Билса - арахнодактилия, контрактуры суставов кистей рук, ки-фосколиоз, аномальная форма ушных раковин, генерализованная остео-пения, которая может способствовать развитию деформаций различных отделов опорно-двигательного аппарата. Для данного заболевания патология сердечно-сосудистой системы и органа зрения не характерны. При редком атипичном варианте синдрома Марфана найдены мутации в одном из генов коллагена I типа (COL1A2). Марфаноподобный фенотип наблюдается также при прогрессирующей диафизарной дисплазии 1 типа Камурати-Энгельманна, вызванной мутациями в гене Tgfpi - TGFBL Второй тип синдрома Марфана связан с мутациями в гене рецептора 2 Tgfpi - TGFBR2. Мутации в гене TGFBR1 найдены у больных синдромом Фурлонга - марфаноподобной болезнью II типа, сочетающейся с краниосиностозом и умственной отсталостью. Аллельным вариантом каждого из этих двух заболеваний является синдром аневризмы аорты Лоеса-Диетза. Этот синдром часто путают с синдромом Марфана, так как оба заболевания имеют перекрывающиеся спектры клинических проявлений. В таблице 1 представлены варианты наследственных скелетных дисплазии с марфаноподобным фенотипом.
Таблица 1. Некоторые аллельные варианты синдрома Марфана и наследственные скелетные дисплазии с марфаноподобным фенотипом
Дата добавления: 2014-11-24 | Просмотры: 1922 | Нарушение авторских прав |