|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Миодистрофия Дюшенна/Беккера
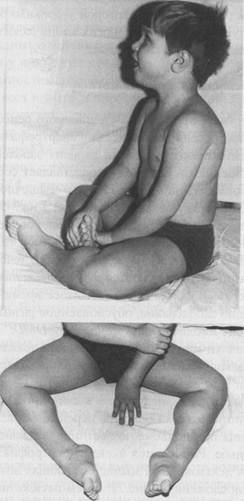
Другим известным примером Х-сцепленного рецессивного заболевания является прогрессирующая псевдогипертрофическая мышечная дистрофия Дюшенна/Беккера. Первый вариант заболевания, подробно описанный Г. Дюшенном в 1868 году, представляет собой одну из наиболее частых и злокачественных форм нервно-мышечной патологии детского возраста. Частота заболевания составляет 1 на 3-5 тысяч новорожденных мальчиков. В 1955 году П. Беккер описал более мягкий вариант Х-сцепленной прогрессирующей мышечной дистрофии, встречающийся с частотой 1 на 20- 25 тысяч лиц мужского пола. В течение долгого времени шли дискуссии о том, являются эти заболевания одной или разными нозологическими формами. В настоящее время убедительно доказано, что это одно заболевание, обусловленное разными мутациями в одном и том же гене миодистрофии Дюшенна - DMD. Первые признаки миодистрофии Дюшенна появляются в возрасте 2-7 лет. Для большинства больных характерна задержка раннего моторного развития. При начале ходьбы (в возрасте 14 месяцев и старше) отмечаются частые падения, неловкость в движениях, быстрая утомляемость. Постепенно походка становится переваливающейся, затем появляются затруднения при подъеме по лестнице, вставании из положения на корточках, ходьбе. Развивается псевдогипертрофия преимущественно икроножных и дельтовидных мышц, создающая ложное впечатление атлетического телосложения - рис.71. Затем псевдогипертрофия трансформируется в гипотрофию. Патологический процесс носит восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса, спины и проксимальных отделов верхних конечностей. По мере развития заболевания возникают вторичные деформации позвоночника (поясничный гиперлордоз, кифоз, сколиоз), грудной клетки, которая становится седловидной или килевидной, стоп. Формируются «осиная талия», крыловидные лопатки, симптом «свободных надплечий». Постепенно развиваются обездвиженность, рестракции сухожилий, контрактуры суставов. Вместе с уменьшением массы мышц угнетаются рефлексы. Сопутствующим признаком заболевания является кардиомиопатия, которая проявляется в виде гипертрофии левого желудочка и аритмии. Примерно у четверти больных диагностируется олигофрения в степени дебильно-сти. Больные сохраняют способность к ходьбе до 10-12-летнего возраста, после чего передвигаются только с помощью инвалидной коляски. Основной причиной летального исхода в возрасте до 20-25 лет являются интеркуррентные инфекции, которые больной не в силах перенести из-за включения в патологический процесс дыхательной мускулатуры. Прогрессирующая мышечная дистрофия Беккера дебютирует обычно во второй декаде жизни с появления слабости и утомляемости мышц тазового пояса и ног. Одним из ранних симптомов, проявляющихся у значительного числа больных, являются болезненные мышечные спазмы. Симптомы мышечной дистрофии при двух формах заболевания носят сходный характер, но при форме Беккера выражены значительно слабее. Гипертрофическая или дилатационная кардиомиопатия диагностируется у 50-60% больных. Болезнь носит медленно прогрессирующий характер, инвалидизация наступает чаще всего после 40 лет. При этом интеллект, как правило, сохранен. Больные вступают в брак, имеют здоровых детей, работоспособны. Картированию гена DMD в области Хр21.2, а затем и его и идентификации способствовало описание единичных случаев миодистрофии Дюшенна у девочек. Эти девочки оказались носителями редких транслокаций между Х-хромосомой и одной из аутосом, причем таких, которые разрушали ген миодистрофии Дюшенна. Основным продуктом гена DMD в мышцах является структурный стержневидный белок дистрофии, принадлежащий к спектрин/а-актининовому суперсемейству белков цитоскелета. Это полифункциональный белок, обеспечивающий поддержание целостности мембраны мышечного волокна при раундах сокращения-расслабления, а также участвующий в формировании ней-ромышечного синапса. Дистрофии состоит из четырех доменов и располагается на цитоплазматической поверхности мышечной сарколеммы -рис.72. N-концевой домен дистрофина связан с цитоскелетом мышечного волокна. Затем идет самый крупный домен, обеспечивающий гибкость молекулы. Он имеет структуру трехгранного стержня и образован 24 слегка повторяющимися мотивами. За стержневым доменом следуют два очень важные в функциональном отношении домена - цистеин-богатый и С-концевой. В области цистеин-богатого домена формируются кальциевые каналы и осуществляется связь дистрофина, а значит и цитоскелета мышечного волокна с внеклеточным матриксом через трансмембранный комплекс дистрофин-ассоциированных белков, которые, в свою очередь, разделяют на два субкомплекса - саркогликановый и дистрогликановый. В области С-концевого домена располагается синтрофиновый комплекс дистрофин-ассоциированных белков, функции которого особенно важны для формирования нейромышечного синапса. В 65-70% случаев у больных миодистрофией Дюшенна/Беккера диагностируются протяженные внутригенные делеции, затрагивающие несколько соседних экзонов, причем эти делеции характерны для обеих форм заболевания. Различия в их характере заключаются в том, что при миодистрофии Дюшенна делеции сопровождаются сдвигом рамки считывания. В этих случаях дистрофии у больных вообще не образуется. При форме Беккера делеции не нарушают рамку считывания, и дистрофин у больных синтезируется, хотя конечно, он имеет аномалии. Клинические проявления подобных делеции зависят от их протяженности и локализации. Так, например, описан больной с очень мягкой формой миодистрофии Беккера, диагностированной в среднем возрасте (после 40 лет), у которого при молекулярном анализе была выявлена протяженная делеция, затрагивающая более 40% гена. Однако эта делеция была так удачно расположена, что в мутантном дистрофине отсутствовал внутренний участок стержневого домена, в то время как функционально значимые домены 1, 3 и 4 оказались совершенно сохранными. Это и обеспечило такое мягкое течение заболевания.. С другой стороны гораздо меньшие по размеру
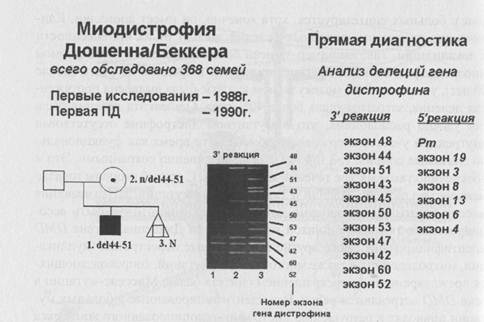
Рисунок 71. Гипертрофия икроножных мышц при миодистрофии Дюшенна. делеции и даже точковые мутации, затрагивающие цисеин-богатый и С-концевые домены дистрофина, могут быть ассоциированы с тяжелой клиникой миодистрофии Дюшенна. В гене DMD идентифицированы также другие внутригенные перестройки (дупликации, микроделеции), а также много нонсенс-мутаций, сопровождающихся преждевременным прекращением синтеза белка. Миссенс-мутации в гене DMD встречаются редко. Все идентифицированные у больных мутации приводят к разрушению дистрофин-ассоциированного комплекса белков, что и служит патогенетической основой для развития мышечной дистрофии при этом заболевании. Молекулярная диагностика делеции в гене DMD проводится с использованием мультиплексной (множественной) ПЦР, что позволяет во многих семьях высокого риска проводить профилактику заболевания на базе пренатальной диагностики. На рис.73 представлен пример пре-натальной диагностики делеции в гене DMD методом мультиплексной ПЦР и указано количество семей больных, обследованных в лаборатории пренатальной диагностики наследственных и врожденных заболеваний ИАГ им. Д. О. Отта РАМН (зав. лаб. чл-корр. РАМН проф. В. С. Баранов). В 30% семей мать больного миодистрофией мальчика не является носителем мутации, а болезнь у сына развивается в результате спонтанного возникновения мутации de novo в её яйцеклетках. Такую ситуацию очень важно диагностировать, так как в этом случае риск повторного рождения больного ребенка может быть значительно меньше и в некоторых случаях не превышает общепопуляционного. Разработаны специальные методы для выявления гетерозиготного носительства мутаций в гене DMD, в частности, иммуногистохимический анализ дистрофина в биоптате мышц
Рисунок 72. Структура дистрофина и дистрофин-ассоциированного комплекса белков. .
Рисунок 73. Диагностика делеций в гене DMD у больных миоди-строфией Дюшенна (данные 2002 г.). Возможна иммунологическая диагностика заболевания с использованием специфических антител на дистрофии. Материалом для исследования в этом случае является образец мышцы, полученной методом биопсии. Серьезную проблему для медико-генетического консультирования представляют случаи гонадного мозаицизма, обусловленного возникновением мутации в первичных половых клетках, то есть на ранних стадиях внутриутробного развития будущей матери. Считается, что 6-7% всех спорадических случаев являются следствием гонадного мозаицизма у матери. При этом оценить величину аномального клона ооцитов не представляется возможным. Эмпирический риск повторного рождения больного ребенка в спорадических случаях миодистрофии Дюшенна и при отсутствии доказательств гетерозиготного носительства мутации у матери достигает 14%, и это делает обоснованным пренатальную диагностику заболевания не только в семейных, но и в спопалических cnv чаях. Дата добавления: 2014-11-24 | Просмотры: 1782 | Нарушение авторских прав |