|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Миодистрофия Дюшенна-Беккера
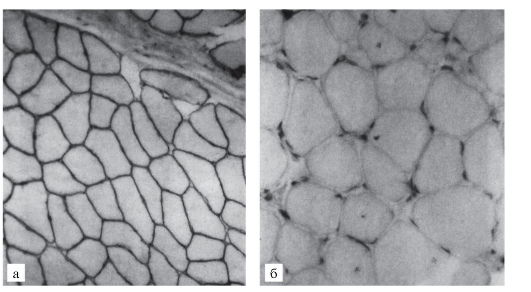
Это одна из частых форм многочисленных наследственных нервномышечных заболеваний. Мышечные дистрофии характеризуются прогрессирующими дегенеративными изменениями в поперечнополосатой мускулатуре без первичной патологии периферического мотонейрона. Миодистрофия Дюшенна-Беккера вызвана мутацией в гене, ответственном за синтез белка дистрофина. Этот белок находится в большом количестве в области сарколеммы, поддерживая, по-видимому, целостность мембраны (рис. 4.29). Структурные изменения в сарколемме приводят к дегенерации цитоплазматических компонентов, усиленному входу Са2+ внутрь волокон, что вызывает гибель миофибрилл. Генетически единая форма миодистрофии Дюшенна-Беккера клинически разделяется на миодистрофию Дюшенна и миодистрофию Беккера.
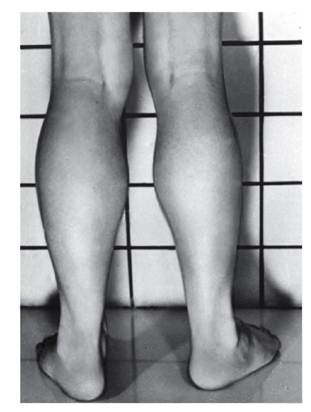
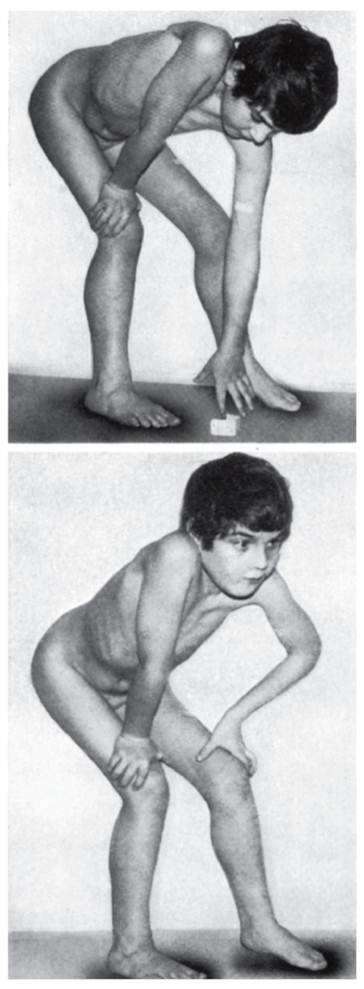
Рис. 4.29. Гистохимическая картина мышц в норме (а) и при миопатии Дюшенна (б) Миодистрофия Дюшенна встречается с частотой 1: 5000 живорожденных мальчиков. Генетически она относится к Х-сцепленным рецессивным летальным нарушениям. Болезнь проявляется рано. Первые симптомы появляются в возрасте до 2 лет: дети позднее начинают ходить, не умеют бегать и прыгать. Симптомы становятся выраженными в 2-3-летнем возрасте. Это изменения походки («утиная» походка), псевдогипертрофия икроножных мышц (рис. 4.30). Процесс атрофии мышц постепенно приобретает восходящее направление: мышцы бедра тазовый пояс плечевой пояс руки. Наблюдается псевдогипертрофия не только икроножных мышц, но и ягодичных, дельтовидных, мышц живота, языка. У детей развиваются поясничный лордоз, крыловидность лопаток (рис. 4.31). Из наклоненного положения больные с трудом распрямляются, опираясь на колени (рис. 4.32). Атрофический процесс развивается и в сердце (кардиомиопатия). Острая сердечная недостаточность - причина летальных исходов. Нарушается моторика ЖКТ. Обнаруживаются вторичные изменения в костной системе. Интеллект у больных детей снижен. Корреляции между тяжестью мышечного дефекта и степенью снижения интеллекта нет. На самой последней стадии атрофия (слабость) захватывает мышцы лица, глотки и дыхательные мышцы. Больные умирают на 2-3-м десятилетии жизни. Из биохимических показателей для миодистрофии Дюшенна наиболее характерен резко повышенный (в 10-100 раз) уровень креатинфосфокиназы в сыворотке крови. Активность этого фермента повышена в первые дни внеутробной жизни и, возможно, даже во внутриутробном периоде. При клинической картине миодистрофии Дюшенна у девочек следует исключить моносомию по Х-хромосоме (синдром Тернера). Редкая возможность миодистрофии Дюшенна у девочек с кариотипом 46,ХХ не исключается из-за инактивации Х-хромосомы с нормальным аллелем во всех (или почти всех) клетках на ранних стадиях развития (16-32-клеточная бластоциста).
Рис. 4.30. Псевдогипертрофия икроножных мышц
Рис. 4.32. Трудности при распрямлении после наклона Гетерозиготные носительницы миодистрофии Дюшенна могут иметь субклинические симптомы: увеличенные икро- ножные мышцы, повышенную утомляемость при физической нагрузке, изменения электромиограммы, увеличение уровня креатинфосфокиназы в крови. Более или менее выраженные симптомы миодистрофии Дюшенна отмечаются у 70% гетерозиготных носителей. Миодистрофия Беккера - доброкачественная форма нервномышечных болезней. Частота миодистрофии Беккера у новорожденных мальчиков составляет 1:20 000. По клиническим симптомам миодистрофия Беккера напоминает миодистрофию Дюшенна, но в менее выраженной форме. Начало болезни не ранее 10-15 лет, течение мягкое, больные сохраняют работоспособность в возрасте 20-30 лет. Фертильность не снижена. Нарушения интеллекта и кардиомиопатии не отмечаются. Активность креатинфосфокиназы заметно повышена, но не в такой степени, как при миодистрофии Дюшенна. Мягкая (или доброкачественная) форма миодистрофии Беккера объясняется тем, что, в отличие от миодистрофии Дюшенна, когда полностью прекращается синтез дистрофина, при миодистрофии Беккера синтез дистрофина детерминируется, но либо вырабатывается мало белка, либо продуцируется аномальный дистрофин. В настоящее время разрабатывается терапия миодистрофии Дюшенна-Беккера: генная (заместительная), клеточная (миобласты, стволовые клетки), аминогликозидная терапия, применение ингибиторов протеосом и др. Генетика миодистрофии Дюшенна и миодистрофии Беккера хорошо изучена (еще с 1930-х годов). Это типичный пример Х-сцепленного рецессивного наследования. Ген дистрофина локализован в коротком плече Х-хромосомы, он уже клонирован и секвенирован. Это самый длинный ген из всех изученных. В нем более 2х106 пар оснований (более 60 интронов); длина мРНК 16 000 пар оснований. В связи с большой длиной в гене часто наблюдаются перестройки, ведущие к мутациям. Доля свежих мутаций среди всех случаев миодистрофии Дюшенна-Беккера составляет 30%. Поскольку ген секвенирован, то молекулярно-генетическая диагностика геми- и гетерозиготных состояний возможна почти во всех семьях. Миодистрофия Дюшенна-Беккера в 60-70% случаев вызывается большими делециями гена дистрофина (по меньшей мере одного экзона). Дупликация больших сегментов обнаруживается у 10% пациентов. Остальные 30% случаев обусловлены точковыми мутациями (нуклеотидные замены и делеции одного нуклеотида). Сообщается также о случаях малых вставок и делеций (2-5 пар нуклеотидов). Делеции в экзонах размером более 5 нуклеотидов встречаются крайне редко. Недавно был идентифицирован ген-модификатор миодистрофии Беккера - ген миогенного фактора-6 (MYF6), локализованный в хромосоме 12q21. Мутации в этом гене совместно с легкими изменениями гена дистрофина ведут к тяжелой клинической картине миодистрофии (о клиническом полиморфизме мышечной дистрофии Дюшенна см. также в статье О.В. Напалковой и др. на компакт-диске). Дата добавления: 2015-02-06 | Просмотры: 1095 | Нарушение авторских прав |