|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Патогенез болезни на молекулярном уровнеВ зависимости от контролируемого конкретным геном продукта и от характера его нарушения при мутации соответствующим образом развертывается патогенез болезни на молекулярном уровне. Если в результате мутации будет вырабатываться избыточное количество продукта, то патогенез болезни в целом будет обусловлен усиленной генной активностью. Существование таких вариантов можно предполагать, но обнаружен он лишь в единичных формах наследственных болезней. Пример такой генной мутации - мутация в гене FII, приводящая к усиленному синтезу протромбина. При другом варианте патологического эффекта мутантного гена синтезируется аномальный белок. За этим следуют нарушения той системы (клетки, органа), функции которой обеспечиваются нормальным белком. Эти нарушения первоначально развертываются на молекулярном уровне. Примером такого варианта патогенеза болезни может быть серповидно-клеточная анемия. В результате замены урацила на аденин в кодоне GUA синтезируется цепь молекулы глобина с глютамином, заменившим валин. Замены одной аминокислоты оказывается достаточно, чтобы изменить функциональные свойства гемоглобина (пониженную растворимость, повышенную полимеризацию). Такой гемоглобин уже не может выполнять кислородакцепторную функцию и кристаллизуется при недостатке кислорода, а эритроциты приобретают серповидную форму (отсюда и название болезни), склеиваются, тромбируют капилляры и т.д. (рис. 4.2). Третий вариант патологического эффекта мутантного аллеля - отсутствие выработки первичного продукта. Этот вариант, очевидно, встречается наиболее часто. Естественно, что в этих случаях нарушается тот или другой процесс нормального биохимического гомеостаза. Это выражается в накоплении токсичных продуктов-предшественников (рис. 4.3). На схеме представлены
Рис. 4.2. Мазок крови больного серповидно-клеточной анемией (б) по сравнению с нормой (а). Патология: серповидные эритроциты, пойкилоцитоз, анизоцитоз, склеенные эритроциты. Пояснения в тексте
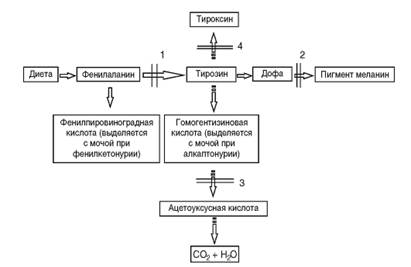
Рис. 4.3. Биохимические «блоки» при наследственных нарушениях обмена аминокислот: 1 - фенилкетонурия; 2 - альбинизм; 3 - алкаптонурия; 4 - врожденная недостаточность тироксина (дисгормоногенез) результаты наследственных нарушений аминокислот фенилаланина и тирозина. При фенилкетонурии в крови накапливаются фенилаланин и продукты его патологического метаболизма (1), поскольку он из-за отсутствия фенилаланингидроксилазы не превращается в тирозин. Нарушение обмена тирозина приводит к патологии образования меланина (2) или тироксина (4). Недостаточность синтеза оксидазы гомогентизиновой кислоты (сущность мутации в этом гене) ведет к накоплению гомогентизиновой кислоты в крови (3), которая из-за высокой концентрации откладывается в хрящах и клапанах сердца. В конечном счете с возрастом это приводит к артритам и порокам сердца. Возможны и другие (обходные) пути обмена, часто также с патологическим исходом. В результате отсутствия первичного продукта гена может задерживаться какой-либо важный процесс, постоянно осуществляющийся в организме. Так, мутации генов, детерминирующих синтез ферментов репарации ДНК, приводят к невозможности восстановления постоянно возникающих нарушений в структуре ДНК, что обусловливает развитие злокачественных новообразований (пигментная ксеродерма, атаксиятелеангиэктазия). Известен и 4-й вариант первичного патологического эффекта мутантного аллеля - выработка уменьшенного количества нормального первичного продукта (β-талассемия, акаталазия). Патогенез таких заболеваний различен, поскольку наряду с нормальным путем обмена веществ будут протекать и патологические процессы. Выше были описаны общие закономерности патогенеза генных болезней на молекулярном уровне на примерах нарушения обмена веществ. Тот же самый принцип патогенеза (мутантный аллель - патологический первичный продукт) действует и для генов морфогенетического контроля, мутации в которых приводят к врожденным порокам развития (полидактилия, синдромы Холта- Орама, Крузона, Нунан, Лоренса-Муна, Меккеля, Робертс, Эллиса-Ван-Кревельда, Гольтца: рис. 4.4, 4.5). Начальное звено врожденного порока развития связано с нарушением дифференцировки клеток. Запрограммированные в геноме дифференцировка клеток, а затем и органогенез осуществляются путем смены процессов активации и выключения определенных генов в строго ограниченных временных (по отношению к онтогенезу) промежутках (транскрипционные факторы). Если первичный продукт морфогенетического гена аномальный, то необходимая для дальнейшего правильного развития органа дифференцировка клеток не последует. Естественно, что морфогенетических генов много, они действуют в разные периоды онтогенеза. Соответственно мутации в них будут приводить к специфическим врожденным порокам развития. Дата добавления: 2015-02-06 | Просмотры: 1156 | Нарушение авторских прав |