|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
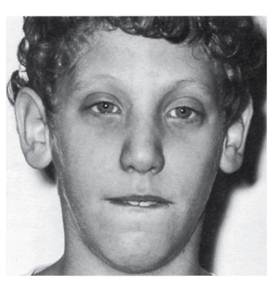
Синдром умственной отсталости с ломкой Х-хромосомойСиноним: синдром Мартина-Белл. Ранее эту болезнь называли синдромом олигофрении с маркерной Х-хромосомой. Синдром Мартина-Белл - одна из наиболее часто встречающихся (после болезни Дауна) форм умственной отсталости. Популяционная частота заболевания составляет 1:2000-5000 живорожденных. Больных мальчиков в 2-3 раза больше, чем девочек, мальчики болеют тяжелее. Частота носительства от 1:165 до 1:1540. Внешность больных неспецифична, но определенное диагностическое значение имеют удлиненное лицо, высокий выступающий лоб, макро- и долихоцефалия, гипоплазированная средняя часть лица (особенно в сравнении с выступающими и часто увеличенными щеками), выступающий подбородок (рис. 4.33). Нёбо обычно дугообразное, губы толстые, нижняя губа часто вывернута. Отмечаются также увеличенные оттопыренные ушные раковины, большие кисти и стопы. Одно из типичных проявлений синдрома - макроорхидизм. При рождении у детей яички нормальных размеров, но с наступлением пубертатного периода они существенно увеличиваются в результате избыточного роста соединительной
Рис. 4.33. Лицо подростка с синдромом Мартина-Белл ткани и накопления жидкости. Однако половая активность таких больных минимальна, к половой жизни способны единицы. Возможны повышенная растяжимость кожных покровов, слабость связочного аппарата суставов (что приводит к самопроизвольным вывихам, обычно пальцев кисти), плоскостопие. У многих больных формируется пролапс митрального клапана. Все эти симптомы обусловлены врожденной дисплазией соединительной ткани. Умственная отсталость, типичная для синдрома Мартина-Белл, как правило, умеренная или выраженная, хотя у 10-15% больных обнаруживаются глубокая олигофрения и еще у такого же процента больных - мягкая умственная отсталость. Большинство пациентов социально адаптированы, выполняют несложную физическую работу. Психологи оценивают их как контактных и доброжелательных. Лишь в тяжелых случаях таких больных приходится помещать в специализированные интернаты. Неврологические симптомы включают мышечную гипотонию и в отдельных случаях судороги. Пороки развития и нарушения других органов сравнительно редки; это расщелины нёба, нистагм, страбизм, птоз, катаракта, кривошея в сочетании со сколиозом, кифоз, дефект межпредсердной перегородки. Помимо определенного комплекса клинических аномалий, синдром Мартина-Белл сопровождается характерной цитогенетической картиной: ломкостью в дистальной части длинного плеча Х-хромосомы (в зоне Xq), что внешне напоминает «спутник» длинного плеча (рис. 4.34). Эта ломкость выявляется лишь при культивировании лимфоцитов в условиях дефицита фолиевой кислоты, поэтому для обнаружения ломкости нужно либо использовать культуральные среды, лишенные фолиевой кислоты, либо вводить в культуральную среду антагонисты фолиевой кислоты. Однако даже и при этих условиях ломкость Х-хромосомы выявляется не во всех клетках (до 60%). Долго считали, что синдром наследуется по Х-сцепленному рецессивному типу. Однако в родословных отмечались случаи тяжелой болезни у женщин и легкой у мужчин. У женщин-гетерозигот отмечали некоторое снижение интеллекта, чаще оцениваемое как пограничная умственная отсталость. У некоторых из них в небольшом проценте клеток обнаруживали ломкую Х-хромосому. Таким образом, наследование синдрома Мартина-Белл не укладывалось в строгие рамки Х-сцепленного рецессивного наследования. Более того, в родословных отмечалась антиципация. Этиология этого заболевания была выяснена с помощью методов молекулярно-генетического анализа. Обнаружена экспансия нестабильных тринуклеотидных повторов (CGG) в 5'-нетранслируемой области гена FMR1 (fragile mental retardation). В норме в этом гене число повторов варьирует от 6 до 42. Частота экспансии премутационных триплетных повторов в гене FMR1 в полную мутацию в оогенезе является функцией длины премутационного аллеля, который имеется у гетерозиготной женщины (рис. 4.35). Хромосомы, в которых имеется 50-200 повторов, считают премутацией. Для этого состояния характерны незначительные проявления: задержка развития, признаки аутизма, атаксия. В следующем поколении число повторов может увеличиться (экспансия) до 1000 и более, что и обусловит выраженную клиническую картину, зависящую от числа повторов. Огромное влияние оказывает также цитозиновое метилирование повторов. Такое состояние называют полной мутацией. Соответственно если женщина унаследовала большое число повторов, то она будет больной. Обнаружены еще два синдрома [FXTAS (синдром тремора и атаксии, ассоциированный с ломкой Х-хромосомой) и FRAXE (синдром ломкой Х-хромосомы)], обусловленные динамическими мутациями в локусе Xq27.3, но эти гены (FMR1 и FMR2) являются причиной болезни намного реже. При этом клиническая картина сходна с синдромом Мартина-Белл, но не идентична.
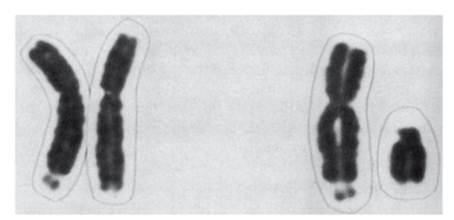
Рис. 4.34. Ломкая Х-хромосома при синдроме Мартина-Белл: слева - набор Х-хромосом женщины (одна хромосома ломкая); справа - мужской набор (ломкая Х-хромосома) Диагноз ставят на основании клинической картины и по результатам клинико-генеалогического и цитогенетического исследований. Наиболее точный метод - молекулярно-генетическая диагностика, в том числе с использованием методов оценки метилирования. В настоящее время возможна пренатальная диагностика данного синдрома. Этиотропной терапии пока не существует. Дата добавления: 2015-02-06 | Просмотры: 1215 | Нарушение авторских прав |