|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
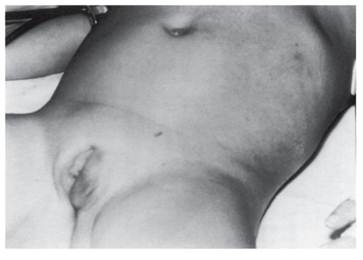
Простая вирильная формасопровождается прогрессирующей вирилизацией, ускоренным соматическим развитием, повышенной экскрецией гормонов коры надпочечников. У новорожденных девочек при кариотипе 46,ХХ отмечается маскулинизация различной выраженности - от умеренной гипертрофии клитора (рис. 4.28) до полного срастания губно-мошоночных складок с формированием мошонки и пениса. Внутренние половые органы сформированы правильно по женскому типу. У мальчиков вирильная форма адреногенитального синдрома при рождении обычно не распознается (гениталии нормальные). Диагноз устанавливают лишь на 5-7-м году жизни при появлении первых признаков преждевременного полового развития. Рис. 4.28. Вирилизация наружных половых органов у девочки с адреногенитальным синдромом Неклассическая форма дефицита 21-гидроксилазы, или поздняя форма, вызвана снижением активности фермента на 40-80% по сравнению с нормальными значениями и клинически проявляется только в подростковом возрасте. У девочек наблюдаются умеренное увеличение клитора, раннее развитие молочных желез, более раннее закрытие зон роста скелета, нарушение менструального цикла, гирсутизм, аспе, у женщин - нарушение менструального цикла и бесплодие. Симптомом избытка андрогенов у мальчиков может быть ускоренное закрытие зон роста костей, олигоспермия с последующим снижением фертильности у мужчин. У детей обоего пола наиболее частым симптомом неклассической формы заболевания является раннее оволосение на лобке и под мышками (адренархе).
Латентная форма не имеет клинических проявлений, но в сыворотке крови умеренно повышен уровень предшественников кортизола. Некоторые авторы не выделяют такие состояния в отдельную форму болезни. Средняя частота дефицита 21-гидроксилазы составляет 1: 13 500 новорожденных (в Швеции - 1: 9800, в США - 1: 15 981, в Европе - 1: 14 970, в Японии - 1: 18 000), а в некоторых изолированных популяциях она в 5-10 раз выше. В России частота адреногенитального синдрома составляет 1: 8000-1: 10 000. Адреногенитальный синдром относят к группе заболеваний, подлежащих просеивающей диагностике у новорожденных. Чем раньше установлен диагноз адреногенитального синдрома, тем более эффективной оказывается терапия глюкокортикоидами и минералокортикоидами, хотя не все проявления заболевания поддаются коррекции. Лучше всего лечится неклассическая форма. Более целесообразно проведение пренатальной диагностики и пренатального лечения дефицита 21-гидроксилазы в семьях, где оба родителя гетерозиготны и, как правило, уже имеют больного ребенка. Это позволяет родителям решить вопрос о сохранении беременности. Если беременность решено сохранить, то проводят внутриутробную терапию глюкокортикоидами (девочка избавляется от хирургической коррекции гениталий). У мальчиков своевременно диагностируется сольтеряющая форма, диагностика которой в постнатальном периоде часто задерживается. Ген стероид-21-гидроксилазы (CYP21A2) локализован в коротком плече хромосомы 6 (6р21.3), это часть гена цитохрома Р450. Его мутантные формы вызывают недостаточность 21-гидроксилазы. При разных клинических формах адреногенитального синдрома, как правило, имеются свои спектры мутаций. Однако причины широкой вариабельности клинических симптомов адреногенитального синдрома полностью не ясны. Строгой корреляции между генотипом и фенотипом (сольтеряющая или простая вирильная форма) нет. Одни и те же мутации, идентифицированные на молекулярногенетическом уровне, имеют разные клинические проявления. Так, было проведено ДНК-обследование более 200 больных с адреногенитальным синдромом с описанием степени вирилизации женских наружных половых органов; секреции андрогенов в ответ на стимуляцию адренокортикотропным гормоном (АКТГ); реакции на бессолевую диету путем оценки дефицита альдостерона и потери соли. Больные были разделены на 26 групп с полностью идентичными мутациями. Оказалось, что в половине групп фенотипические проявления и генотип не коррелируют. Однако неклассическая форма дефицита 21-гидроксилазы в 90% случаев обусловлена двумя миссенс-мутациями в гене цитохрома QYP21. Среди причин клинического полиморфизма наряду с особенностями мутаций можно назвать также и модифицирующее влияние других генов. Так, тяжелая сольтеряющая форма адреногенитального синдрома ассоциируется с антигенами системы HLA: A3, Bw47, DR7. Дата добавления: 2015-02-06 | Просмотры: 902 | Нарушение авторских прав |