|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
ЭТИОЛОГИЯ. Генные болезни - разнородная по клиническим проявлениям группа заболеваний, обусловленных генными мутациями
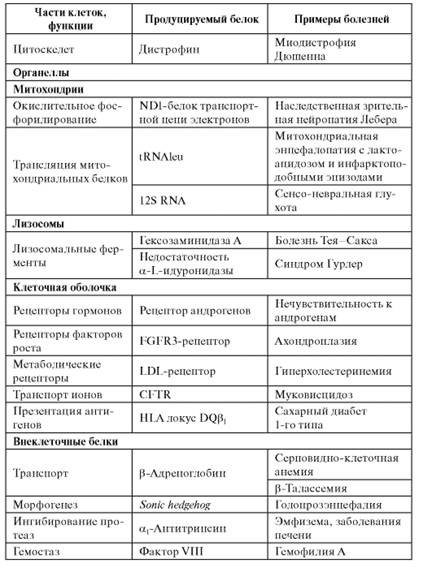
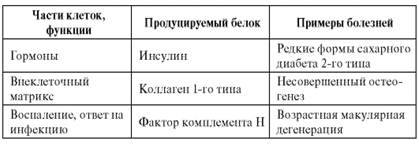
Генные болезни - разнородная по клиническим проявлениям группа заболеваний, обусловленных генными мутациями. Основой для объединения их в одну группу служат этиологическая генетическая характеристика и соответственно закономерности наследования в семьях и популяциях. Коль скоро мутации в индивидуальных генах являются этиологическим фактором генных болезней, то закономерности их наследования соответствуют менделевским правилам расщепления в потомстве, т.е. формальная генетика генных наследственных болезней ничем не отличается от «поведения» в семьях любых менделирующих признаков. «Поведение» некоторых патологических генов может отклоняться от менделевско-моргановских правил в связи с фенотипическими эффектами (летальность, стерильность). Необходимо, однако, сразу сделать пояснения в отношении содержания понятий «генные мутации» и «менделирующая наследственность» у человека. Во-первых, согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом, можно говорить о многообразии видов мутаций в одном и том же гене, которое является причиной наследственных болезней. У человека описаны все типы генных мутаций, обусловливающие наследственные болезни: миссенс, нонсенс, сдвиг рамки считывания, делеции, вставки (инсерции), нарушения сплайсинга, увеличение числа (экспансия) тринуклеотидных повторов. Любой из этих видов мутаций может вести к наследственным болезням. Даже одна и та же генная болезнь может быть обусловлена разными мутациями одного и того же гена. Например, в гене муковисцидоза описано около 300 вызывающих болезнь мутаций (всего их более 1500) следующих типов: делеции, миссенс, нонсенс, сдвиг рамки считывания, нарушения сплайсинга. Для гена фенилкетонурии известно более 30 патологических мутаций (миссенс, нонсенс, делеции, нарушения сплайсинга). Во-вторых, современная генетика, принимая в полной мере менделизм, делает поправки в определенных случаях. К ним относятся условность понятий о доминантности и рецессивности, неоднородность проявления аллеля, унаследованного от отца или матери (импринтинг), сложное взаимодействие генов, гонадный мозаицизм и т.д. Более того, установлено, что мутации в разных частях одного гена ведут к различным болезням. Например, мутации в разных частях RET-онкогена ведут к 4 клинически разным наследственным болезням: двум формам полиэндокринного аденоматоза (ZA) и (ZB), семейной медуллярной тиреоидной карциноме, семейной болезни Гиршпрунга. Мутации, вызывающие наследственные болезни, могут затрагивать структурные, транспортные и эмбриональные белки, ферменты. Белковые классы, ассоциированные с моногенными болезнями, имеются фактически во всех составных элементах клетки (табл. 4.1). Таблица 4.1. Примеры классов белков, ассоциированных с моногенными болезнями
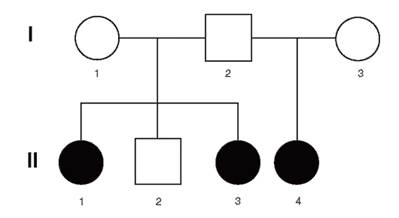
Существует несколько уровней регуляции синтеза белков: претранскрипционный, транскрипционный, трансляционный. Можно предположить, что на всех этих уровнях, обусловленных соответствующими ферментативными реакциями, могут проявляться наследственные аномалии. Если принять, что у человека примерно 30 000 генов и каждый ген может мутировать и контролировать синтез белка с другим строением, а многим генам свойственно еще явление альтернативного сплайсинга, то, казалось бы, должно быть не меньшее число наследственных болезней. Более того, по современным данным, в каждом гене может возникать до нескольких сотен вариантов мутаций (разные типы в различных участках гена). На самом деле более чем для 50% белков изменения генетической природы (первичная структура) приводят к гибели клетки, и мутация не реализуется в наследственную болезнь. Такие белки называются мономорфными. Они обеспечивают основные функции клетки, консервативно сохраняя стабильность видовой организации этой клетки. При рассмотрении генных болезней как менделирующих признаков организма речь идет о так называемых полных формах, т.е. формах, обусловленных гаметическими (в зародышевых клетках) мутациями. Это могут быть новые или унаследованные от предыдущих поколений мутации. Следовательно, в этих случаях патологические гены присутствуют во всех клетках организма. Однако теоретически можно представить возможность появления и мозаичных, а не только полных форм, подобно хромосомным болезням. Любые мутации, в том числе и генные, могут возникать на ранних стадиях дробления зиготы в одной из клеток, и тогда индивид будет мозаичен по данному гену. В одних клетках у него будет функционировать нормальный аллель, в других - мутантный или патологический. Если эта мутация доминантная, то она проявится в соответствующих клетках и, очевидно, приведет к развитию менее тяжелой формы болезни. Если возникшая мутация в одной из клеток на ранних стадиях развития зародыша рецессивная, то ее эффект проявится только у гетерозиготы. Вероятность появления двух рецессивных мутаций в одном и том же локусе гомологичных хромосом в одной соматической клетке чрезвычайно мала. Проблема мозаичных форм генных болезней и в генетическом, и в клиническом плане исследована недостаточно. Частота возникновения мозаичных форм не может быть высокой, поэтому выявлять их трудно. Современные молекулярно-генетические методы позволяют диагностировать мозаицизм на клеточном или тканевом уровне. В одной и той же ткани обнаруживают клетки, несущие разные генотипы по изучаемой патологической мутации. Соматические мутации, появляющиеся на ранних стадиях развития организма, дают больший эффект, чем мутации на поздних. В последние годы соматический мозаицизм был доказан при 30 генных болезнях, среди которых нейрофиброматоз I типа, миотоническая дистрофия, миодистрофия Дюшенна, гемофилия А и В, синдром Альпорта, синдром Марфана, синдром андрогенной нечувствительности, тубероз- ный склероз и др. Соматический мозаицизм был обнаружен также при злокачественных новообразованиях (колоректальный рак и рак предстательной железы). С мозаичными формами генных болезней не следует путать мозаицизм гонад. Мозаицизм гонад - частный случай органного мозаицизма, возникающего на более поздних стадиях эмбрионального развития в процессе органогенеза. Мозаицизм гонад у клинически здорового индивида может обусловить несколько случаев рождения детей с полной формой доминантной наследственной болезни. На рис. 4.1 приведена родословная здоровых родителей (французская семья), у которых трое из четверых детей больны ахондроплазией. Ахондроплазия - аутосомно-доминантное заболевание с полной пенетрантностью гена. Клиническая и рентгенологическая диагностика этой болезни (в частности, в упомянутой выше семье) не вызывает сомнений. Объяснить семейные случаи можно гонадным мозаицизмом у отца, поскольку больные дети родились в двух его браках. Возможно еще одно объяснение подобных случаев: болезнь возникла в результате премутации в одном из аллелей этого гена у
Рис. 4.1. Родословная семьи с 3-мя случаями ахондроплазии в одном поколении от двух браков: I: 1 - рост 163 см; 2 - рост 166 см; 3 - рост 164 см; клинических и радиологических признаков ахондроплазии не имели; II: 1 - родилась от неродственного брака (мать 17 лет, отец 30 лет). Ахондроплазия заподозрена при рождении, позже подтверждена; 2 - здоровый мальчик; 3 - больная ахондроплазией. Диагноз установлен после рождения; 4 - во втором браке отца диагноз ахондроплазии у его ребенка был установлен внутриутробно на 7-м месяце [сначала с помощью ультразвукового исследования (УЗИ), затем рентгенографически]. При рождении диагноз подтвержден родителя, которая реализуется в мутацию при прохождении через мейоз. Однако в гене ахондроплазии премутантных состояний пока не обнаружено. Современные молекулярно-генетические исследования показали, что родительский мозаицизм (в том числе гонадный) ответствен не менее чем за 5-15% случаев доминантных и Х-сцепленных рецессивных болезней. Мозаицизм гонад у здоровых родителей убедительно доказан в случаях рождения детей (по соответствующим генам) с несовершенным остеогенезом, синдромом Элерса-Данло, гемофилией (факторы VIII и IX). В связи с многообразием мутаций в одном и том же гене возникает вопрос об этиологической зависимости клинической картины болезни от характера мутаций. Ответ на этот вопрос неоднозначен и пока не всегда ясен. Определенно можно сказать, что в большинстве случаев такой зависимости нет, хотя в некоторых случаях она присутствует. Объяснение этому можно найти в первичных механизмах развития генных болезней, т.е. в первичных эффектах мутантных аллелей. С клинико-генетической точки зрения эти аллели называют патологическими для отличия от других мутантных состояний этого же гена, которые ведут к биологическим межиндивидуальным вариациям без патологических проявлений признака. Первичные эффекты мутантных аллелей могут проявляться в 4-х вариантах: отсутствии синтеза полипептидной цепи (белка); синтезе аномальной по первичной структуре полипептидной цепи (белка); количественно недостаточном синтезе полипептидной цепи (белка); количественно избыточном синтезе полипептидной цепи (белка). Независимо от характера изменений первичного продукта гена эффект мутаций может выражаться в разных вариантах нарушения функций: - Потеря функции белка в результате либо ингибирования процессов транскрипции/трансляции, либо изменения его структуры и функциональных свойств. - Появление новой функции. При мутациях такого типа у мутантного белка наряду с нормальной функцией появляются новые цитотоксические свойства, приводящие к гибели клеток. - Доминантный негативный эффект проявляется тогда, когда первичный продукт мутантного аллеля ингибирует функцию нормальных белков. - Изменение дозы гена (делеции или дупликации) может приводить к нарушению пространственной структуры молекулярного продукта. На основе первичного эффекта мутантного аллеля развертывается весь сложнейший патогенез генной болезни, проявляющийся в разнообразных фенотипических эффектах или клинической картине. Результатом действия патологической мутации (фенотипический эффект) может быть, прежде всего, летальность на ранних стадиях развития зародыша, до имплантации. Механизмы такой летальности еще не выяснены, но ее существование у человека не вызывает сомнений. Это проявляется в виде несостоявшегося зачатия (имплантации) у фертильных женщин при нормальной половой жизни. У молодых женщин зачатие наступает в среднем через 3 мес регулярной половой жизни (без контрацепции). Примерно 50% несостоявшихся зачатий обусловлены гибелью зиготы по генетическим причинам (генные, хромосомные и геномные мутации). Если развитие эмбриона с патологической генной мутацией не остановилось на ранних стадиях, то фенотипические эффекты в зависимости от вовлеченного гена и характера мутации формируются в виде 3 вариантов: дисморфогенеза (врожденных пороков развития), нарушенного обмена веществ, смешанных эффектов (дисморфогенеза и аномального обмена веществ) Влияние патологических мутаций начинает реализовываться в разные периоды онтогенеза - от внутриутробного до пожилого возраста. Большая часть патологических мутаций проявляется внутриутробно (до 25% всей наследственной патологии) и в допубертатном возрасте (45%). Еще 20% проявляется в пубертатном и юношеском возрасте, и лишь 10% моногенных болезней развивается в возрасте старше 20 лет. Для понимания природы генных болезней очень важно иметь представление о том, что клиническая картина заболевания может сформироваться вследствие включения разных патогенетических звеньев, которые могут быть обусловлены фенотипическими эффектами мутаций разных генов. Следовательно, в одну группу будут включены разные с генетической точки зрения заболевания (мутации в разных локусах). Такие случаи называются генокопиями. Наряду с этим, хотя и редко, могут встречаться фенокопии генных болезней. Это те случаи, при которых повреждающие внешние факторы, действующие, как правило, внутриутробно, вызывают болезнь, по клинической картине в общих чертах сходную с наследственной. Противоположное состояние, когда при мутантном генотипе индивида в результате средовых воздействий (лекарства, питание и т.п.) болезнь не развивается, называют нормокопированием. Понятия о гено- и фенокопиях помогают установить правильный диагноз, а также более точно определить прогноз здоровья пациента или вероятность рождения больного ребенка. Понимание принципов нормокопирования дает врачу возможность предупредить развитие болезни у ребенка, унаследовавшего патологический ген. Дата добавления: 2015-02-06 | Просмотры: 1062 | Нарушение авторских прав |