|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
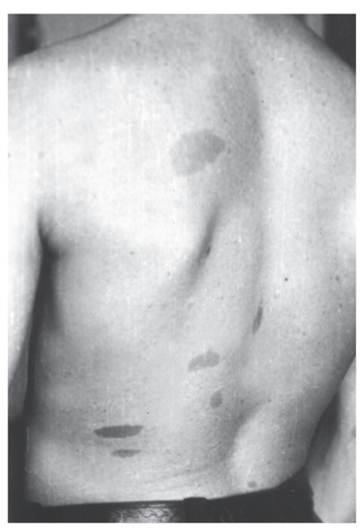
Нейрофиброматоз (болезнь Реклингхаузена)Это тяжелая полисистемная болезнь с аутосомно-доминантным типом наследования. Наиболее тяжело поражается нервная система, поэтому болезнь считают неврологической. Термин «нейрофиброматоз» охватывает по меньшей мере две болезни: нейрофиброма- тоз I типа и нейрофиброматоз II типа, сначала считавшиеся двумя формами одного и того же заболевания (периферический нейрофиброматоз и центральный нейрофиброматоз). Это яркий пример генетической гетерогенности наследственных болезней. Ниже будет описан только нейрофиброматоз I типа. Симптоматика нейрофиброматоза I типа разнообразна, в патологический процесс вовлекается несколько систем, что подтверждает плейотропный эффект гена. Диагноз нейрофиброматоза I типа можно установить при наличии не менее двух из перечисленных ниже признаков, но при условии, что они не являются симптомами какойлибо другой болезни. - Светло-коричневые пигментные пятна (рис. 4.7).
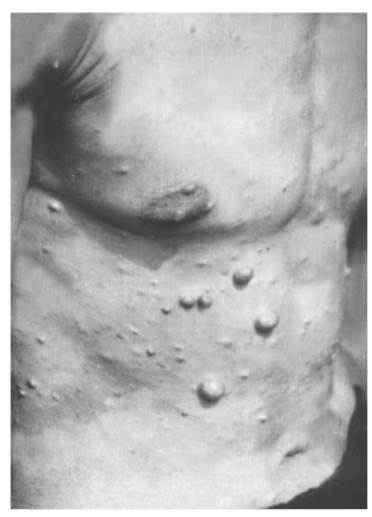
Рис. 4.7. Типичные светло-коричневые пятна у молодого мужчины с нейрофиброматозом I типа У детей их должно быть не менее 5, а диаметр пятен не менее 5 мм. У взрослых число пятен должно быть не менее 6, а диаметр пятен не менее 15 мм. Для выявления пятен необходимо хорошее освещение. Пигментные пятна появляются обычно к 3 годам жизни, их число увеличивается с возрастом. Они встречаются у 95% больных. - Решающий признак - две нейрофибромы любого типа и более или одна плексиформная нейрофиброма (по данным анамнеза или клинического обследования) (рис. 4.8). Нейрофибромы могут возникать в любом участке тела, захватывая кожные нервы, часто располагаются по ходу нервных стволов, иногда захватывают крупные нервы и нервные сплетения (плексиформные нейрофибромы). В месте локализации нейрофибром больные часто ощущают зуд, жжение и боль. У одного больного могут быть тысячи нейрофибром, а их масса может достигать 15 кг и более, если своевременно не сделано их иссечение. У детей таких нейрофибром мало. Их число увеличивается с возрастом, особенно у женщин при беременности. К 30-летнему возрасту нейрофибромы отмечаются у 95% больных. - Множественные, похожие на веснушки пигментные пятна в подмышечной ямке (рис. 4.9), паховой области, на других участках тела со складками. Они обычно возникают в детстве, их число трудно определить. Пигментные пятна обнаруживают у 80% больных. - Костные изменения (дисплазия крыла клиновидной кости, врожденное искривление или утончение длинных трубчатых костей, ложный сустав).
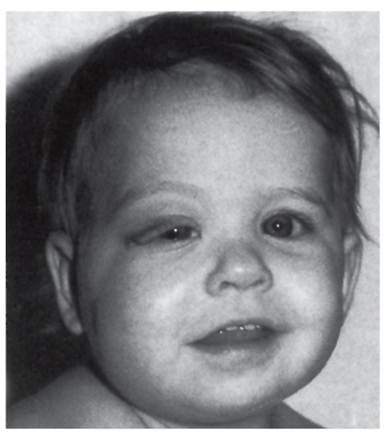
Рис. 4.10. Плексиформная орбитальная нейрофиброма у ребенка Дисплазия глазницы сочетается с плексиформной нейрофибромой глазницы (рис. 4.10). - Глиома зрительного нерва. Эта опухоль характерна для нейрофиброматоза I типа. С помощью КТ и МРТ (магнитнорезонансной томографии) обнаруживается утолщение зрительного нерва. Глиома зрительного нерва протекает обычно бессимптомно, но иногда может вызывать ухудшение зрения, косоглазие, зрачковые аномалии, проптоз и гипоталамическую дисфункцию. - Узелки Лиша (два и более) на радужной оболочке. Узелки представляют собой гамартомные новообразования и не влияют на зрение. После полового созревания узелки Лиша наблюдаются практически у всех больных. Для обнаружения узелков глаза осматривают с помощью щелевой лампы. - Нейрофиброматоз I типа по приведенным выше критериям у родственника I степени родства (родитель, сибс, потомок). Нейрофиброматоз I типа относится к полностью пенетрантным аутосомно-доминантным болезням, поэтому нейрофиброматоз I типа у родственника можно использовать как диагностический критерий. У большинства больных диагноз очевиден уже к 3 годам. Течение заболевания прогрессирующее, с очень большим размахом клинической картины. Наряду с описанными выше симптомами (они либо врожденные, либо зависят от возраста) у 20-30% детей наблюдаются когнитивные нарушения (трудности в обучении). Наиболее опасными проявлениями становятся опухоли - иногда из-за злокачественности, иногда из-за места расположения (черепные нервы, малый таз, ЖКТ). Неблагоприятное течение нейрофиброматоза I типа отмечается у 30% больных. Наряду с основными симптомами у больных нейрофиброматозом часто встречаются осложнения: низкий рост у 25-35% пациентов, плексиформные нейрофибромы у 20%, сколиоз у 10%, головные боли у 20%, нейрофиброматоз 1-ассоциированные злокачественные футлярные опухоли периферических нервов у 7-12%, глиома зрительного нерва у 7%. К более редким осложнениям нейрофиброматоза I типа относятся: эпилепсия у 1-2% пациентов, гидроцефалия у 2%, псевдоартроз у 3%, стеноз почечной артерии у 1-2%, ксантогранулемы - 1-2%, феохромоцитома - менее чем у 1%. Патогенез заболевания пока недостаточно изучен, поскольку трудно объединить разнообразнейшие проявления: пигментные пятна, костные изменения, когнитивные нарушения и т.д. Осложнения могут возникать в любой системе организма, в состав которой входят ткани, происходящие из эктодермы, мезодермы и нервной трубки. Фенотипические проявления нейрофиброматоза I типа значительно различаются даже у членов одной семьи. Однако известно, что ключевую роль играют шванновские клетки, которые в результате соматической мутации теряют гетерозиготность. Так как этот ген является геном-супрессором опухолей, то после потери функции обоих аллелей развиваются опухоли и заболевание манифестирует. Больные нейрофиброматозом I типа встречаются в практике врачей многих специальностей. Пациенты обращаются в первую очередь к дерматологам с жалобами на косметический дефект в виде пигментных пятен и нейрофибром. Наиболее часто больные нейрофиброматозом I типа являются пациентами невропатологов и нейрохирургов. Определенная часть таких больных лечится у хирургов и ортопедов. Генетика нейрофиброматоза I типа на генеалогическом уровне была ясна уже в начале прошлого столетия. В последние годы она изучена до уровня гена. Локус нейрофиброматоза I типа расположен в коротком плече хромосомы 17q11.2. Величина гена - 350 000 пар оснований, в нем 59 экзонов. Ген полностью секвенирован. Он относится к группе генов-супрессоров опухолей, чем и объясняются его опухолевые эффекты. Обнаружено более 500 мутаций (транслокации, делеции, вставки, точковые замены). Однако корреляция клинической картины с типом мутаций еще не установлена. Также обнаружены гены-модификаторы, в настоящее время идет их изучение. Первичный продукт гена назван нейрофибромином. Распространенность нейрофиброматоза I типа равна примерно 1:3500-5000 новорожденных, одинакова у обоих полов, у всех рас и этнических групп. Аутосомно-доминантный тип наследования не нарушается ни в одной популяции. Доля спорадических случаев составляет 50-70%. Частота мутаций по этому гену высокая: 1 мутация на 10 000 гамет на поколение. Лечение нейрофиброматоза I типа в основном хирургическое или симптоматическое. В настоящее время идет разработка патогенетической терапии (антифиброзные средства). Дата добавления: 2015-02-06 | Просмотры: 1266 | Нарушение авторских прав |