|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Клеточный уровень патогенеза генных болезнейПатогенез генных болезней не заканчивается на молекулярном уровне даже в первичных звеньях. Для многих болезней главное звено патогенеза - клетка. Во всех генетических процессах клетка - дискретная самостоятельно регулируемая единица, и в ней осуществляются все процессы реализации генетической информации (транскрипция, трансляция, синтез белка). Это - общебиологическая аксиома. Клеточный уровень патогенеза генных болезней означает, что в определенных типах клеток разыгрываются основные патологические процессы, присущие конкретной нозологической форме. Клетка как бы не выпускает из себя патологические явления, а принимает на себя удар первичного патогенного эффекта гена. Точкой приложения первичного действия мутантного гена являются отдельные структуры клетки, разные при различных болезнях (лизосомы, пероксисомы, мембраны, митохондрии). Патогенетические процессы на клеточном уровне развертываются при болезнях накопления (или лизосомных) в связи с нарушением активности лизосомных ферментов. Так, накопление в клетках, а затем и в основном межклеточном веществе, гликозаминогликанов (мукополисахаридов) приводит к развитию тяжелых заболеваний - мукополисахаридозов. Причина избыточного содержания полимеров - гликозаминогликанов - заключена в отсутствии их деградации в лизосомах. Нарушение деградации гликозаминогликанов связано с дефектами в группе специфических
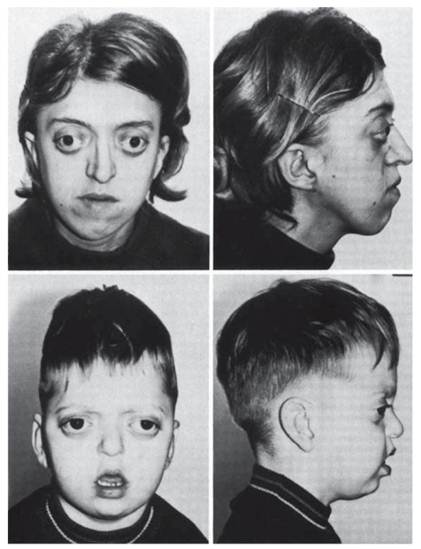
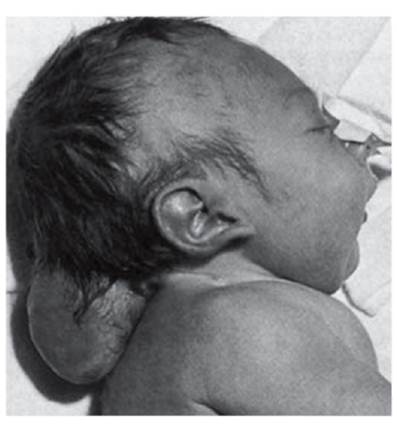
ферментов, катализирующих весь цикл деградации. Более подробную информацию о клинической картине, диагностике и лечении мукополисахаридозов см. в статье С.В. Михайловой с соавт. «Мукополисахарозы: дифференциальня диагностика и лечение» на компакт-диске. Другим примером болезней накопления могут служить гликогенозы. В клетках печени и мышц накапливаются полимеры гликогена, которые не подвергаются деградации даже тогда, когда организму необходима глюкоза в крови. Патогенез гликогенозов принципиально такой же, как и мукополисахаридозов. В клетках печени и мышц отсутствует определенный фермент (их уже известно много), который участвует в цикле расщепления гликогена до глюкозы. Другие внутриклеточные структуры - пероксисомы - также могут быть точкой приложения первичного действия мутантного гена. В этих случаях развиваются так называемые пероксисомные болезни. Описано уже 18 нозологических форм. Основное патологическое звено при всех пероксисомных болезнях локализовано в пероксисомах в виде биохимических нарушений, обусловленных генными мутациями. Биохимическая сущность многих пероксисомных болезней уже раскрыта на уровне мутантных ферментов. Клинически болезни проявляются в виде множественных врожденных пороков развития, в целом сходных при разных нозологических формах (множественных черепно-лицевых дисморфиях, катаракте, кожных складках на шее, почечных кистах и др.). Пероксисомные болезни - пример наследственных болезней обмена, при которых множественные пороки развития объясняются молекулярным дефектом. Различают 3 группы пероксисомных болезней: генерализованные с измененным числом пероксисом (пример - цереброгепаторенальный синдром, или синдром Целлвегера); с неизмененным числом пероксисом и нарушением нескольких биохимических функций (пример - целлвегерподобный синдром); с неизмененным числом пероксисом и нарушением единственной биохимической функции (пример - болезнь Рефсума).
Мембраны, так же, как и структуры клеток, могут быть ключевыми элементами патогенеза генных болезней. Так, отсутствие специфических белковых молекул-рецепторов на клеточной поверхности, связывающих ЛПНП, приводит к семейной гиперхолестеринемии. Синдром полной нечувствительности к андрогенам (синоним: синдром тестикулярной феминизации) вызывается мутациями в Х-сцепленном гене, который кодирует синтез внутриклеточного рецептора андрогенов. Отсутствие чувствительности клеток к андрогенам приводит к развитию женского фенотипа при хромосомном наборе XY. У таких больных, несмотря на женское строение наружных половых органов, имеются семенники в брюшной полости и нормальный уровень андрогенов в крови. Клиника витамин-D-резистентного рахита (аутосомно-доминантное заболевание) обусловлена дефектом рецепторов 1,25-ди- гидроксихолекальциферола. При муковисцидозе нарушается регуляция транспорта хлоридов через мембраны эпителиальных клеток. Такая регуляция в норме осуществляется белком-продуктом гена, названным кистофиброзным трансмембранным регулятором (CFTR). Одни мутации в гене CFTR ведут к снижению синтеза данного белка из-за незавершенности процессинга РНК, другие - к качественным изменениям мембранных хлорных каналов. Одна первичная биохимическая аномалия (нарушение транспорта хлоридов) обусловливает возникновение мультиорганного патологического процесса (прогрессирующее поражение дыхательных путей, хронические синуситы, недостаточность экзокринной секреторной функции поджелудочной железы, стерильность у мужчин). Клеточный уровень патогенеза генных болезней может проявляться не только в конкретных органеллах, но и в виде нарушения скоординированности функций клетки. Так, мутации, затрагивающие области онкогенов, ведут к снятию контроля размножения клеток (репрессия антионкогенов) и соответственно к злокачественному росту (наследственный рак толстой кишки, ретинобластома). Клетка может быть главным звеном при реализации патогенеза на молекулярном уровне. Так, прекращение синтеза мышечного белка дистрофина при мутациях в соответствующем гене приводит к постепенной деградации мышечных клеток. Это спусковой крючок патогенеза тяжелой наследственной болезни - миопатии Дюшенна. Дата добавления: 2015-02-06 | Просмотры: 1173 | Нарушение авторских прав |