|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Миотоническая дистрофияСинонимы: болезнь Штайнерта, дистрофическая миотония. Миотоническая дистрофия - аутосомно-доминантное многосистемное заболевание с сильно вариабельной экспрессией гена, обусловливающей клинический полиморфизм по началу заболевания и тяжести течения. Главные клинические проявления: миотония, мышечная слабость, катаракты, аритмии сердца, облысение со лба, нарушенная толерантность к глюкозе, умственная отсталость. Мышечные судороги особенно выражены в руках, челюстях, языке (в виде фибрилляции). Одновременно постепенно усиливается мышечная слабость в связи с дегенерацией отечных мышечных клеток и атрофией волокон. Миотония и мышечная слабость у пациентов сочетаются с нарушением речи и глотания. Начальные признаки миотонической дистрофии различны. Миотония сначала выявляется только при специальном тестировании. Мышечные подергивания и слабость обычно асимметричны. В первую очередь в патологический процесс вовлекаются лицевые и височные мышцы (миотоническое лицо), затем шейные, плечевые, бедренные мышцы (рис. 4.11, 4.12). В настоящее время известно 2 типа миотонической дистрофии. Миотоническая дистрофия 1-го типа составляет около 98% миотонической дистрофии и характеризуется началом мышечной слабости от дистальных мышц к проксимальным. При миотонической дистрофии 2-го типа мышечная слабость развивается, наоборот, от проксимальных к дистальным отделам.
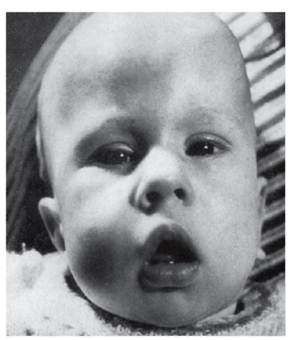
Рис. 4.11. Врожденная форма миотонической дистрофии (мышечная гипотония, амимичное лицо, рот треугольной формы - миотоническое лицо)
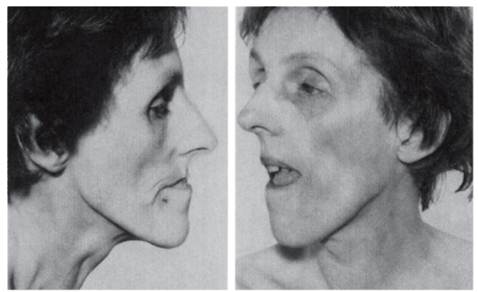
Рис. 4.12. Взрослая пациентка с миотонической дистрофией (птоз, слабость лицевых мышц, атрофия жевательных мышц) Наряду с нервно-мышечными симптомами при миотонической дистрофии отмечаются катаракта (очень ранний симптом), гипогонадизм (атрофия семенников), аменорея, дисменорея, кисты яичника, облысение со лба, изменения проводимости сердца с аритмией, абдоминальные симптомы (на почве холелитиаза), прогрессирующая умственная отсталость. Тяжесть клинических проявлений очень сильно различается даже в пределах одной семьи. Начало миотонической дистрофии возможно от пренатального периода до возраста 50-60 лет. Выделяют 4 формы заболевания (в зависимости от возраста манифестации): врожденную, юношескую, классическую (20-30 лет) и минимальную (50-60 лет). Это объясняется различиями в числе тринуклеотидных повторов в локусе миотонической дистрофии (см. ниже). Смерть при миотонической дистрофии наступает в возрасте 50-60 лет (при классической форме) вследствие пневмонии, сердечных осложнений или других интеркуррентных заболеваний. Частота болезни может различаться в этносах и популяциях. Эффект родоначальника описан у канадцев французского происхождения. В Израиле распространенность болезни в среднем равна 1: 6369 с различиями между общинами: у евреев-ашкенази 1: 17 544, у евреев-сефардов 1: 5000, у евреев-йеменитов 1: 2114. Обобщенно распространенность миотонической дистрофии можно оценить как1: 7500-10 000. Генетика миотонической дистрофии хорошо изучена на генеалогическом, формально-генетическом и молекулярно-генетическом уровнях. При миотонической дистрофии 1-го типа у пациентов всех стран обнаружена одна и та же мутация в гене протеинкиназы мышечной дистрофии (символ гена DMPK), локализованном в хромосоме 19q13.2-19q13.3. Суть мутации - экспансия (увеличение числа) нестабильных CTG-повторов в 3'-нетранслируемой области гена. В норме число CTG-повторов колеблется от 5 до 30. При миотонической дистрофии этот показатель значительно увеличивается и составляет 50-2000 и более. Обнаружена корреляция между тяжестью и числом тринуклеотидных повторов. Чем больше повторов, тем раньше начинается заболевание и тяжелее протекает болезнь. Клиническая картина у гомозигот более тяжелая. При миотонической дистрофии 2-го типа найдена другая мутация - в гене цинко- вых пальцев (ZNF9), локализованном в хромосоме 3q21.3. Мутация представляет собой нестабильную экспансию CCTG-тетраплета с повторами от 75 до 11 000. Во многих семьях с миотонической дистрофией в нескольких поколениях отмечается антиципация, т.е. более тяжелая манифестация болезни и начинающаяся в более молодом возрасте в каждом последующем поколении. Этот признак описан для миотонической дистрофии давно и рассматривался в 1940-х годах как статистический артефакт. Однако сведения о молекулярном дефекте указывают на возможность увеличения числа триплетов в поколениях. Описаны семьи с более чем тремя поколениями с миотонической дистрофией: в 1-м поколении - только катаракты, во 2-м поколении - умеренная слабость мышц, в 3-м поколении - врожденная форма. При миотонической дистрофии выражен импринтинг. Пациенты, рожденные от больных матерей, имеют более тяжелую форму болезни с более ранним началом, чем пациенты, рожденные от больных отцов. Врожденная форма миотонической дистрофии наблюдается только у детей от больных матерей. Механизм импринтинга выяснен: экспансия триплетов происходит в мейозе у женщин, а при сперматогенезе она отсутствует. Уменьшение длины мутантного повтора (почти до нормы) у потомков с легкой клинической картиной или бессимптомным заболеванием наблюдается при передаче гена от отца. Одним из объяснений этого может быть селекция против длинных аллелей в мужском гаметогенезе. Дата добавления: 2015-02-06 | Просмотры: 1633 | Нарушение авторских прав |