|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
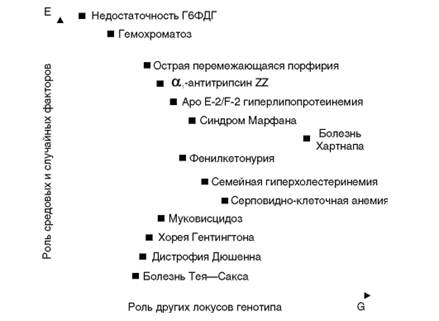
Клинический полиморфизм и его причиныКлиническая генетика всегда опиралась в своих принципах на закономерности, установленные экспериментальной генетикой. Это в полной мере относится к анализу клинического полиморфизма. В 1934 г. Н.В. Тимофеев-Ресовский в статье «Связь между геном и внешним признаком (феноменология проявления генов)» писал «...лишь первый шаг к генетической физиологии развития, а именно к так называемой феноменологии проявления генов. Этим я обозначаю расчленение и классификацию всеобщих явлений в чудовищно многогранной и изменчивой области проявления самых различных наследственных признаков». Он обратил внимание и проиллюстрировал на конкретных экспериментальных материалах «общие феномены проявления генов», среди них такие, как гетерогенные гены, полифенные (плейотропные) гены и константно и вариабельно повторяющиеся гены. Он проявил интерес к концептуальности и практическому использованию таких знаний «прежде всего в области наследственной патологии человека». Спустя почти 40 лет В. Маккьюсик эти феномены проявления генов у человека, не меняя сути в их интерпретации, назвал «принципами клинической генетики»: клинический полиморфизм, генетическая гетерогенность и плейотропизм. Еще раньше в России, как уже отмечалось, С.Н. Давиденков, практикуя в клинике нервных болезней, в 1930-е годы ими широко пользовался. Одна из основных и наиболее старых аксиом клинической медицины сводится к тому, что болезнь любой этиологии (инфекционной, травматической, алиментарной, гормональной и др.) проявляется неодинаково у разных индивидов, поэтому нужно лечить не болезнь, а больного. В ряде случаев клиническая картина одного и того же заболевания варьирует от стертых форм до тяжелейших клинических проявлений. Формирование клинической картины связывают с особенностями действия этиологических факторов (например, вирулентность возбудителя), исходного состояния организма (иммунный статус, обмен веществ), сопутствующих условий (стресс, температура). Кроме того, признается роль врожденных характеристик организма в патогенезе и клинической картине болезней. Казалось бы, можно ожидать более или менее унифицированной клинической картины какой-либо нозологической формы генных болезней, поскольку этиологический фактор для всех больных с этой формой одинаков (мутация в соответствующем гене), а патогенез развертывается на фоне жестко детерминированного контроля генной активности. Такой вывод подсказывал общегенетический взгляд на моногенно детерминируемые события. Однако клиническая практика показала, что симптоматика наследственных болезней различна. При накоплении наблюдений одних и тех же нозологических форм оказалось, что клинический полиморфизм генных болезней выражен не меньше, чем ненаследственной патологии. При многих заболеваниях, достаточно хорошо изученных на клиническом, генетическом и молекулярном уровнях, нет строгой корреляции между генотипом и фенотипом. Неясно, почему заболевания, вызванные строго доказанными одними и теми же мутациями на молекулярном уровне, имеют разные клинические проявления иногда даже у идентичных близнецов. Нужны дальнейшие исследования с анализом генных сетей, активности факторов транскрипции, транспортных белков и других модификаторов экспрессии генов. Клинический полиморфизм генных болезней проявляется в разных сроках начала заболевания, полноте и тяжести симптоматики (глубина патологического процесса), продолжительности болезни, степени инвалидности, толерантности к терапии, в сокращении продолжительности жизни. Вместе с тем следует подчеркнуть, что генные болезни не имеют плавных переходов от нормы к патологии. Даже самая легкая форма болезни обязательно имеет минимальные диагностические критерии. Генетическое правило гласит: нормальный генотип детерминирует нормальный фенотип, а мутантный генотип детерминирует мутантный фенотип (болезнь). Генетической причиной полиморфизма может быть явление взаимодействия главного гена и генов модификаторов (эпистаз, особенности инактивации и дозовая компенсация Х-хромосомы, цитоплазматический геном), с другой стороны - это могут быть и факторы внешней среды, в которых осуществляется развитие индивида. На рис. 4.6 изображено влияние названных двух групп факторов (генетических и внешнесредовых) на фенотипы моногенных болезней. Так, фенилкетонурия в пространстве двух обозначенных координат занимает срединное положение, отражая заметное влияние средовых и случайных факторов, а также эффектов других генов на клинические проявления болезней. В то же время для болезни Тея-Сакса влияние этих факторов менее выражено, а в клинических проявлениях недостаточности Г6ФДГ преобладающим модифицирующим фактором является внешняя среда. К настоящему времени накопился огромный фактический материал по феноменологии клинического полиморфизма отдельных форм и факторам, его определяющим. В первую очередь следует рассматривать значение характера мутации в конкретном локусе для проявления болезни или формирования фенотипа (мутантного). Первично возникшие и унаследованные от предыдущих поколений мутации имеют достаточно сходное фенотипическое проявление, т.е. длительность унаследования мутации не отражается на клиническом полиморфизме генных болезней. Как подчеркивалось выше, десятки и даже сотни разных мутаций (и даже разных типов) в одном и том же локусе ведут к одной и той же болезни. В большинстве случаев характер мутации не определяет клиническую
Рис. 4.6. Потенциальное влияние генетических (G) и негенетических (E) факторов на фенотип некоторых моногенных болезней (по Ч. Скрайверу и П. Уотерс) картину болезни. Фенотип определяет первичный эффект гена (нет продукта или мало продукта). Однако накапливается все больше данных о зависимости фенотипа (клинической картины болезни) от генотипа (разных мутаций в одном и том же локусе). Такие формы болезней, при которых мутации не полностью блокируют выработку первичного продукта, уже известны. Расшифровка корреляций между гено- и фенотипом стала возможной благодаря молекулярно-биологическим исследованиям структуры генов, мутаций и их первичных продуктов. Мутации в одном и том же локусе, ответственные за синтез дистрофина, приводят к двум клиническим формам: миопатии Дюшенна (тяжелой) и миопатии Беккера (легкой). Установлено, что миопатия Дюшенна развивается при полной блокаде, а Беккера - при частичной блокаде синтеза РНК для дистрофина (при миопатии Беккера делеции гена меньшего размера). Сплайсинговые мутации, как правило, не полностью блокируют образование мРНК, поэтому соответствующие формы болезни бывают мягкими по клинической картине и течению. Четкая корреляция между генотипом (характером мутации) и фенотипом (клинической картины болезни) отмечена пока лишь при одном виде мутаций - экспансии тринуклеотидных повторов в гене. Чем больше повторов в мутантном аллеле, тем тяжелее протекает болезнь. Особенно четко это проявляется при синдроме Мартина- Белл и миотонической дистрофии. Поскольку экспансия повторов формируется в мейозе у одного из родителей (у мужчины или женщины при различных болезнях), это обусловливает явление антиципации - более тяжелое течение наследственной болезни в последующих поколениях. До открытия этого типа мутаций (экспансии триплетов) и молекулярно-генетического прослеживания числа повторов в поколениях явление антиципации рассматривалось как артефакт наблюдений. Теперь стало ясно, что феномен антиципации существует. Для него уже обнаружена биологическая основа при небольшой группе болезней. В последнее время появились данные, свидетельствующие о том, что развитие некоторых наследственных заболеваний может быть обусловлено резким изменением числа копий не только тринуклеотидных повторов, но и тандемных повторов ДНК большей протяженности (тетра- и пентануклеотидов, мини- и мегасателлитов), при этом возможно как увеличение, так и, напротив, уменьшение числа копий повторяющегося элемента. Примеры наследственных заболеваний, связанных с так называемыми динамическими мутациями, приведены в табл. 4.2. Большинство болезней, обусловленных нестабильными нуклеотидными повторами, фенотипически (клинически) проявляются главным образом неврологической симптоматикой (атаксии, когнитивные нарушения, деменции, нистагм, паркинсонизм), хотя иногда в болезнь включаются и другие органы (семенники - макроорхидизм, дисплазия соединительной ткани, нарушение сердечной проводимости и др.). Патогенетические механизмы болезней экспансии нестабильных повторов разнообразны. Их можно разделить на три класса. - Класс 1: экспансии некодирующих повторов, вызывающие нарушение функции белков и транскрипции патологического гена преРНК (синдром Мартина-Белл, атаксия Фридрейха). - Класс 2: экспансии некодирующих повторов, вызывающие появление новых свойств РНК (миотоническая дистрофия 1 и 2, синдром тремор/атаксии с ломкой Х-хромосомой). - Класс 3: экспансии кодирующих повторов, приводящие к новым свойствам мутантного белка (хорея Гентингтона, спиномозжечковая атаксия). К настоящему времени обнаружено несколько геновмодификаторов при моногенных заболеваниях человека. Приведем несколько примеров. Хорошо изучено улучшение состояния у гомозигот по мутации, приводящей к β-талассемии, унаследовавших также аллель α-талассемии. Последний выступает как ген-модификатор. Дисбаланс синтеза цепей глобина, обусловленный β-талассемией, улучшается за счет снижения синтеза α-цепей, вызванного мутацией при α-талассемии. Еще один установленный факт - пациенты с муковисцидозом, гомозиготные по наиболее частой мутации, имеют очень вариабельную патологию легких. Показано, что это связано с наличием по меньшей мере одного гена-модификатора. Клиническая картина болезни может зависеть от «дозы» генов (числа аллелей). Так, гомозиготность (два аллеля) при аутосомнодоминантных болезнях определяет более тяжелую клиническую картину, а иногда даже внутриутробную гибель плода (ахондроплазия, синдром Элерса-Данло). Аутосомно-рецессивные болезни проявляются в полной мере при условии гомозиготного состояния по мутантному аллелю. Однако некоторые признаки заболевания могут проявляться и у гетерозигот (один аллель, легкая форма), они усиливаются до клинических проявлений при действии провоцирующих факторов. Например, симптомы кислородной недостаточности (при подъемах на большую высоту) проявляются у гетерозигот по серповидно-клеточной болезни; беременность у гетерозигот по β-талассемии приводит к развитию анемии. Генетические причины клинического полиморфизма могут быть обусловлены не только патологическим геном, но и генотипом в целом, т.е. генотипической средой в виде генов-модификаторов. Геном в целом функционирует как хорошо скоординированная система. Вместе с патологическим геном индивид наследует от родителей комбинации других генов, которые могут усиливать или ослаблять действие патологического гена. В правильности этого положения не приходится сомневаться, хотя реально гены-модификаторы только начинают идентифицировать. Сравнение выраженности клинической картины у членов одной семьи и разных семей показывает, что межсемейные различия больше, чем внутрисемейные, но внутрисемейные тоже существуют. Примером гена-модификатора может служить ген, локализованный в длинном плече хромосомы 1, кодирующий «повсеместный» транскрипционный фактор (upstream stimulatory factor 1, USF1), участвующий в метаболизме углеводов и липидов у больных с семейной комбинированной гиперлипидемией. Показано, что разные аллельные варианты этого гена определяют разный уровень его транскрипции и, как следствие, USF-факторы активируют или подавляют экспрессию многих генов, вовлеченных в липидный (АРОЕ, АРОА1, АРОА5) и углеводный (глюкокиназа, рецептор глюкагона) обмены, а также участвующих в регуляции уровня артериального давления (ренин, ангиотензиноген). Важно, что аллельные варианты USF1 вносят заметный вклад в риск сердечно-сосудистых заболеваний на популяционном уровне. Таким образом, модифицирующая функция гена USF1 в отношении генов, вовлеченных в регуляцию липидного и углеводного обменов, а также уровня артериального давления, делает этот ген ответственным за признаки, составляющие метаболический синдром. Одним из факторов вариабельности фенотипа или разной экспрессивности может быть соматический мозаицизм. На клиническом уровне явление индивидуальной модификации действия патологического гена изучал С.Н. Давиденков на примерах наследственных болезней нервной системы. Многие «мелкие», наследуемые независимо от основного патологического гена, признаки усиливают клинические проявления болезни. Это явный признак взаимодействия генов. С.Н. Давиденков (1947) высказал гипотезу «условного тропизма патологических нервных задатков»: «Помимо своего прямого влияния на развитие нервной системы, патологический задаток обладает еще способностью усиливать эффект от других наследственных факторов, обладающих сходно направленным тропизмом». В развитии генной болезни, как и любого наследственного признака человека, имеет значение не только генотип, но и внешняя среда. Выше рассмотрена роль таких генетических факторов, как характер мутаций патологического аллеля и генотипическая среда (взаимодействия генов) в формировании клинического полиморфизма. В то же время не вызывает сомнений влияние окружающей среды в широком смысле слова на развитие болезни. Этому положению есть много доказательств из клинической практики и специальных исследований. Приведем несколько примеров. Симптоматика фенилкетонурии у ребенка более тяжелая, если во время его внутриутробного развития в рационе матери было много продуктов, богатых фенилаланином. Обострение наследственных миодистрофий наблюдается после стрессов, охлаждений, переутомления. Клиническая картина гемофилии у ребенка усиливается с увеличением у него кровоизлияний от падений и травм. У женщин, больных нейрофиброматозом I типа, резко усиливается рост нейрофибром при беременности. Дата добавления: 2015-02-06 | Просмотры: 3424 | Нарушение авторских прав |