|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
ФИЗИОЛОГИЯ ГЕМОСТАЗА
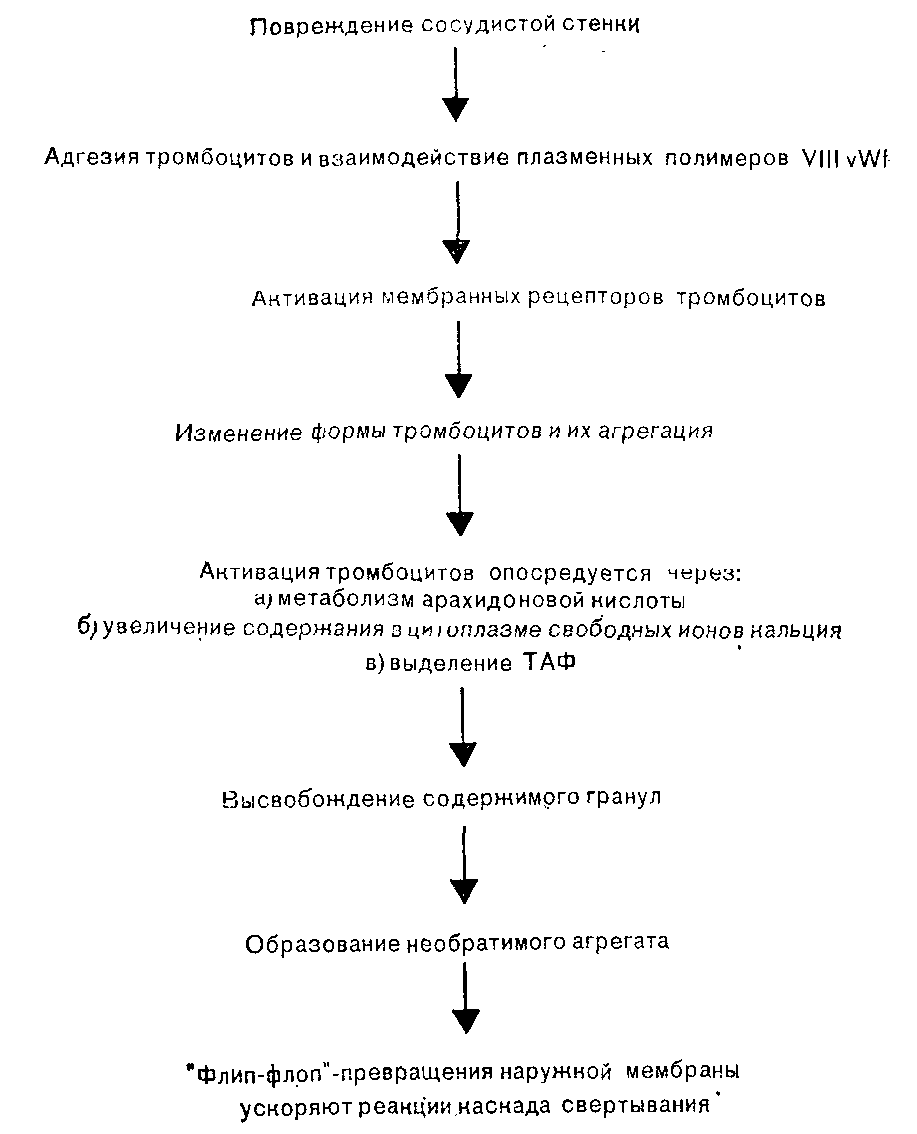
Процесс гемостаза состоит в предупреждении кровотечений посредством поддержания целости замкнутой системы кровеносных сосудов с различным давлением находящейся в них крови, и восстановления проходимости сосуда в случае его закупорки. Гемостатические реакции можно разделить на несколько совпадающих и последовательных событий: локальный сосудистый спазм в месте повреждения, адгезия тромбоцитов к оголенной субэндотелиальной базальной мембране и волокнам коллагена, образование агрегата тромбоцитов или тромба, активация каскада свертывания, приводящая к образованию фибрина, армирующего тромб, и, наконец, активация фибринолитической системы, которая переваривает гемостатический тромб и способствует росту новых клеток сосудистого эндотелия [Sixma, Wester, 1977]. Кроме этого, существует сложная система физиологических ингибиторов и механизмов обратной связи для контроля и ограничения любой активации системы гемостаза, если такая активация избыточна или не соответствует потребностям организма [Bennet, 1977]. Развивающийся вслед за повреждением сосудистый спазм длится меньше одной минуты и, видимо, мало влияет на скорость кровопотери. При этом просвет сосуда суживается не более чем на Уз его исходного диаметра. Механизм сосудистого спазма неясен. По всей вероятности, он связан с нейрогенным сокращением сосудистой стенки и выделением из активированных тромбоцитов различных веществ, таких как серотонин и тромбоксан А2. Интактный сосудистый эндотелий не вызывает сколько-нибудь значительной активации различных компонентов крови. На рис. 7 показаны взаимодействия, возникающие вслед за повреждением сосудистой стенки. При нарушении целости сосудистой стенки обнажаются ее субэндотелиальные структуры, включая коллаген и микрофибриллы базальной мембраны. Циркулирующие тромбоциты взаимодействуют с обнажившимися коллагеновыми волокнами и прилипают к поврежденной поверхности, чему способствуют фибронектин и высокомолекулярные полимеры комплекса фактора VIII, покрывающие поверхность тромбоцитов. В состав комплекса фактора VIII входит фактор VIII Виллебранда (VIHvWf, VIII von Willebrand factor), который участвует в начальной адгезии циркулирующих тромбоцитов к поврежденным субэндотелиальным структурам [Zimmerman, Ruggeri, 1983]. VIII vWf синтезируется в клетках сосудистого эндотелия и мегакариоцитах и присутствует в нормальных тромбоцитах. Белок VIII vWf состоит из субъединиц с молекулярной массой около 200 000—240 000, которые легко образуют димеры, а впоследствии и крупные полимеры с молекулярной массой, достигающей 20 млн. Именно находящиеся в плазме полимеры VIII vWf с молекулярной массой свыше 8 млн необходимы для адгезии тромбоцитов к поврежденной сосудистой стенке.
Рис. 7. Участие тромбоцитов в процессе гемостаза [Vermylen et al., 1983].
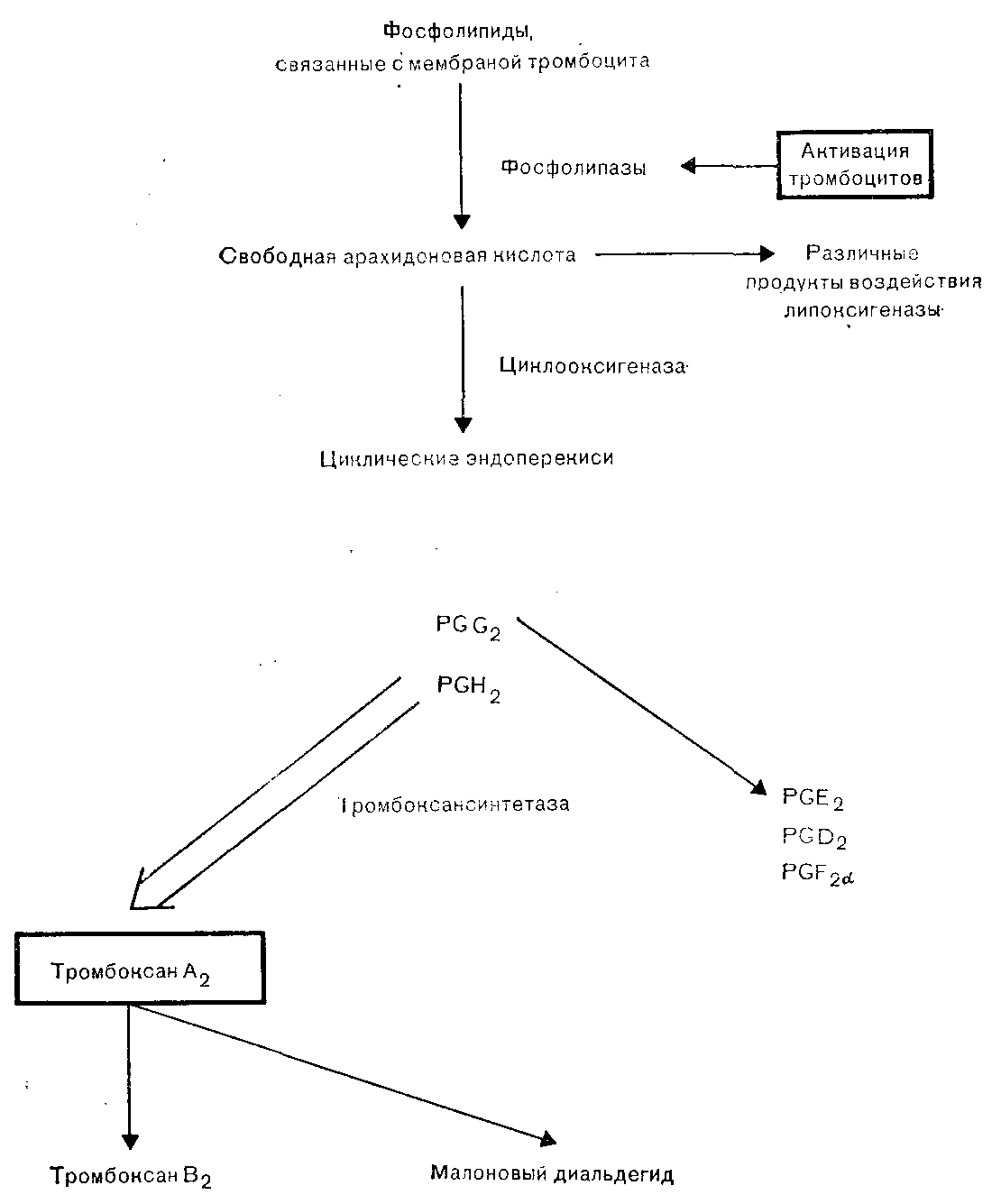
В местах нелинейного тока крови, например на поврежденном участке сосуда или в зоне атеросклеротической бляшки, из локально поврежденных эритроцитов выделяется аденозин-5-ди-фосфат (АДФ), который активирует тромбоциты и индуцирует адгезию [Born et al., 1976]. Циркулирующие тромбоциты представляют собой безъядерные диски, состоящие из трехслойной фосфолипидно-белковой мембраны, субмембранно расположенных кольцевых сократительных филаментов, гранул трех типов и сети канальцев, по которым содержимое гранул может выделяться на поверхность тромбоцита. После адгезии к поврежденному сосудистому эндотелию одного слоя тромбоцитов эти клетки склеиваются между собой и образуют агрегаты. Определенные факторы, взаимодействуя со специфическими поверхностными рецепторами тромбоцитов, вызывают агрегацию, а затем активацию последних. К числу указанных факторов относятся обнаженные коллагеновые волокна, АДФ, высвободившийся из поврежденных эритроцитов и агрегировавших тромбоцитов, адреналин, серотонин, тромбин, а также некоторые метаболиты арахидоновой кислоты, включая тромбоксан А2 (ТХА2). С началом агрегации тромбоциты изменяют свою форму, утрачивая очертания диска и превращаясь в микросферы с многочисленными выступающими псевдоподиями. Агрегация тромбоцитов человека может быть активирована посредством по крайней мере двух, а возможно, и трех независимых, но взаимосвязанных механизмов. Первый механизм активации связан с метаболизмом арахидоновой кислоты [Smith, 1981]. Активация ферментов фосфолипаз приводит к выделению свободной арахидоновой кислоты из фосфолипидов мембраны. Примерно 50% свободной арахидоновой кислоты под действием фермента липоксигеназы вначале превращается в ряд продуктов, включающих различные лейкотриены, которые, по-видимому, играют в регуляции гемостаза очень небольшую роль. Оставшаяся часть арахидоновой кислоты под воздействием фермента циклооксигеназы превращается в циклические эндопероксиды, простагландины G2 и Н2 (PGG2 и PGH2), являющиеся очень лабильными веществами. Затем в результате действия ферментного комплекса тромбоксансинтетазы большинство эндопероксидов быстро превращается в тромбоксан А2 (ТХА2). ТХА2 обладает выраженной биологической активностью, вызывая выделение содержимого гранул тромбоцитов, локальный сосудистый спазм и местную агрегацию других тромбоцитов. Гранулы тромбоцитов делятся на три группы: плотные гранулы, из которых выделяются АДФ, АТФ и серотонин; альфа-гранулы, выделяющие несколько компонентов, в том числе фактор стимуляции гладких мышц, тромбоцитарный фактор 4, обладающий способностью нейтрализовать гепарин, b-тромбоглобулин, фактор VIII RAg, фактор V и фибриноген и лизосомальные гранулы. Высвобождение содержимого разных гранул поддерживает дальнейшее образование агрегатов тромбоцитов. ТХА2 очень лабильный продукт с периодом полужизни in vivo примерно 45 с, он распадается на неактивные вещества: тромбоксан В2 (ТХВ2) и малоновый диальдегид. Небольшая часть циклических эндопероксидов превращается в первичные простагландины, PGE2, PGD2, PGF2a. Метаболизм арахидоновой кислоты в тромбоцитах схематически представлен на рис. 8.
Рис. 8. Метаболизм арахидоновой кислоты тромбоцитов.
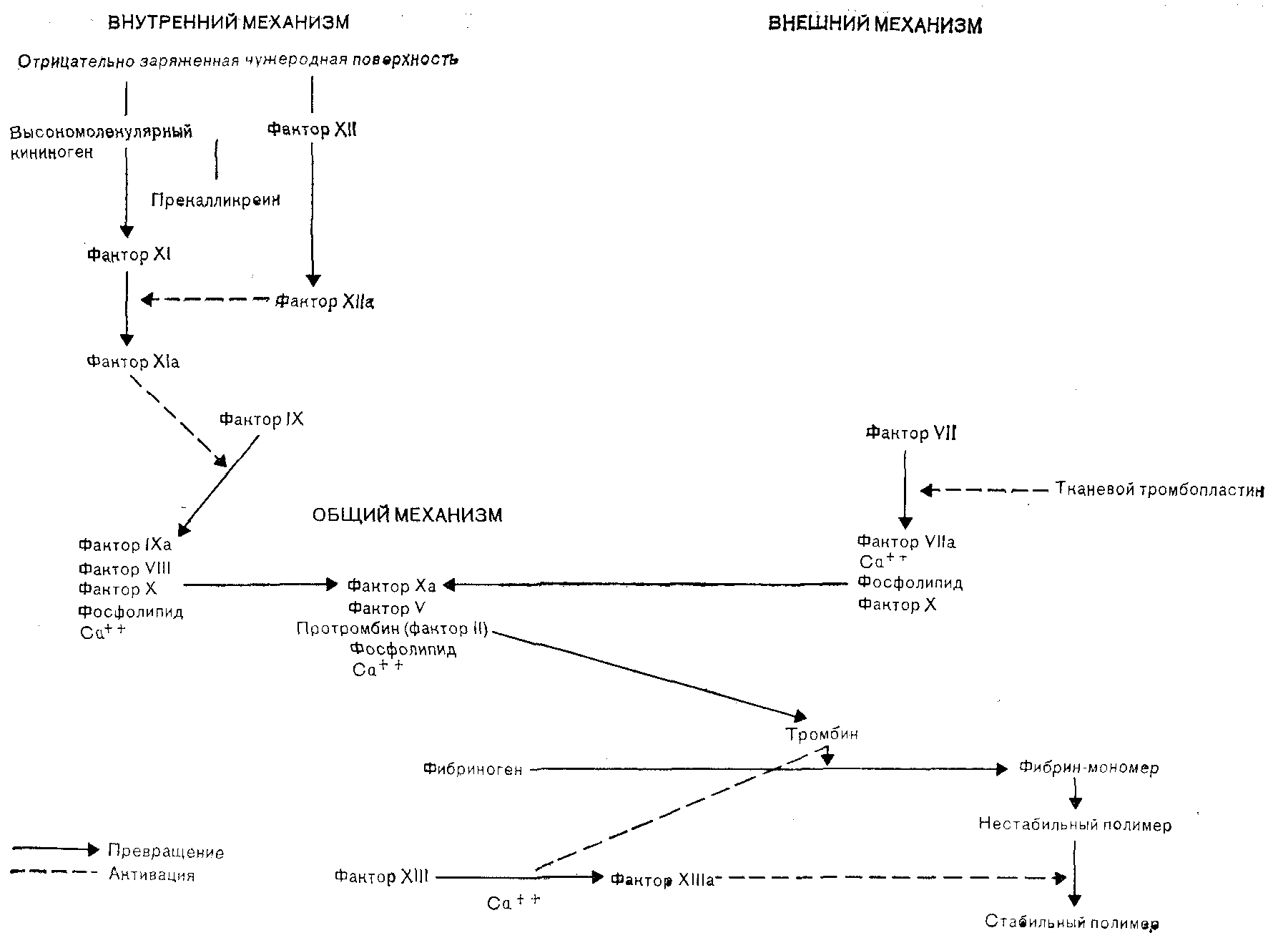
Второй механизм активации тромбоцитов совершенно независим от метаболизма арахидоновой кислоты и образования тромбоксана А2. Различные активаторы тромбоцитов, включая тромбин и коллаген, вызывают внезапное увеличение в цитоплазме этих клеток содержания свободного кальция [White, 1980]. Кальций выделяется из системы плотных трубочек и образует комплекс с кальмодулином. Кальций-кальмодулиновый комплекс действует как кофермент в серии тромбоцитарных реакций. Он вызывает выделение содержимого гранул, высвобождает арахидоновую кислоту из фосфолипидов мембраны, так что она становится доступной для превращения в тромбоксан А2, и активирует актомиозиновую сократительную систему филаментов, расположенных под мембраной тромбоцита. Третий механизм состоит в высвобождении из фосфолипидов тромбоцитарной мембраны лизолецитинового соединения, названного ТАФ (тромбоцит-активирующий фактор), который, по-видимому, активирует тромбоциты независимо от образования тромбоксана А2 и выделения кальция [Chignard et al., 1980]. Действительное значение активации тромбоцитов человека посредством ТАФ изучено недостаточно полно. Вначале тромбоциты образуют относительно рыхлый агрегат, однако вслед за высвобождением содержимого тромбоцитарных гранул формируется необратимый тромб больших размеров. При этом изменяется конфигурация мембраны, вследствие чего значительно ускоряются некоторые реакции свертывания, происходящие на поверхности тромбоцитов [Zwaal, Hemker, 1982]. Механизм свертывания, включающий многокомпонентную ферментную систему проферментов, кофакторов и ингибиторов, активируется при контакте с отрицательно заряженной поверхностью поврежденных эндотелиальных клеток (внутренний путь) и в результате локального выделения тканевого тромбопластина (внешний путь). Процесс свертывания схематически изображен на рис. 9. Сложное взаимодействие фактора XII и высокомолекулярного кининогена, абсорбированных на отрицательно заряженной поверхности эндотелиальных клеток, а также связывание прекалликреина и фактора XI с высокомолекулярным кининогеном ведет к активации фактора XI [Thompson et al., 1977]. Затем следует серия превращений инертных предшественников в активные сериновые протеазы, в результате чего образуется фибрин. Диффузия тканевой жидкости в кровяное русло активирует внешний путь, превращая фактор VII в фактор Vila, который в свою очередь активирует фактор X. Мицеллы фосфолипидов ускоряют две основные реакции свертывания, происходящие на поверхности тромбоцита: во-первых, взаимодействие активированного фактора 1Ха с фактором VIII, что приводит к активации фактора X, и, во-вторых, взаимодействие активированного фактора Ха с фактором V, обеспечивающее образование тромбина из протромбина. Факторы VIII и V не являются сериновыми протеазами, но они способствуют ориентации факторов свертывания на поверхности тромбоцита, усиливая тем самым протеолиз фактора X и протромбина соответственно. Тромбоциты могут также напрямую запускать процесс свертывания, непосредственно активируя фактор XI и фактор X. После образования тромбина фибриноген распадается на мономеры фибрина. Эти мономеры полимеризуются, формируя непрочный фибриновый сгусток, который стабилизируется благодаря действию фактора XIII. Тромбин дополнительно усиливает гемостатический механизм, увеличивая активность факторов VIII и V и вызывая активацию тромбоцитов. Наконец, стабилизированный фибриновый сгусток переплетается с агрегатом тромбоцитов, захватывая некоторое количество эритроцитов, и в месте начального повреждения сосудистой стенки образуется прочный тромб.
Рис. 9. Процесс свертывания крови.
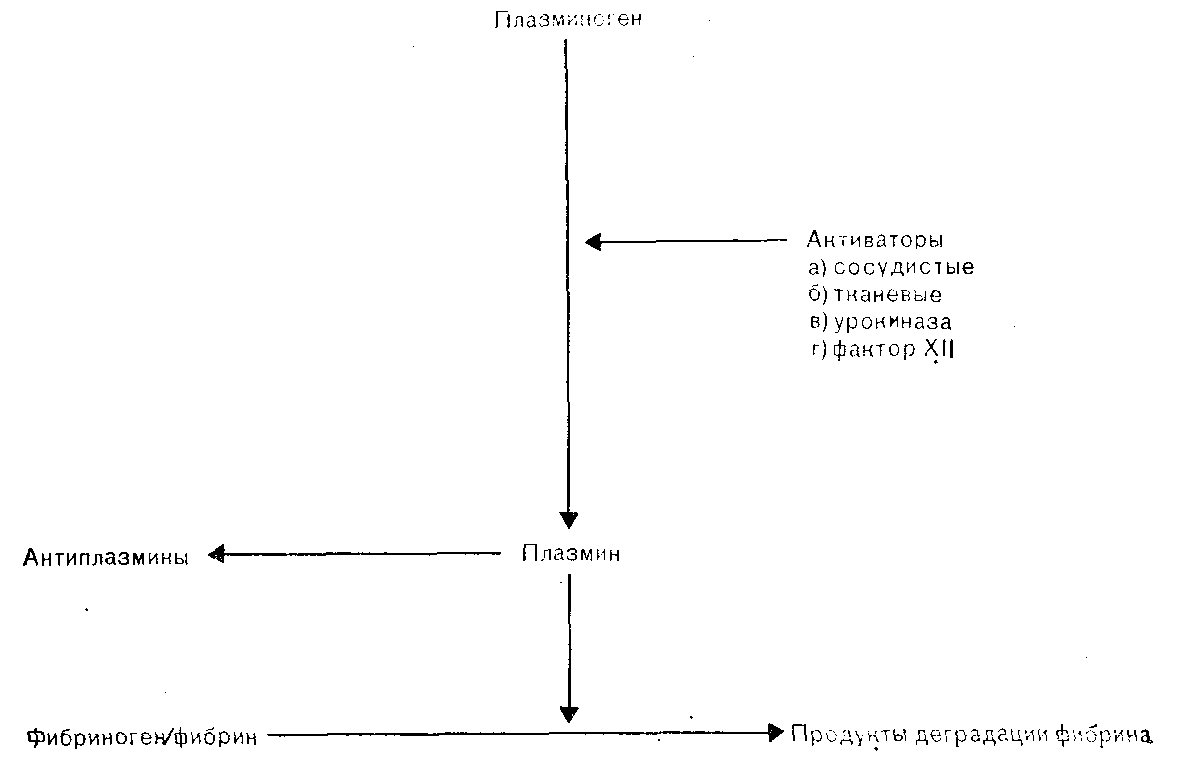
Рис. 10. Фибрииолитическая система [Caffney, 1981].
Активация описанного выше механизма гемостаза должна ограничиваться зоной начального повреждения сосуда, после чего должно происходить заживление дефекта и восстановление целостности эндотелия. Лизис фибринового сгустка зависит от активации фибринолитической ферментной системы, которая схематически изображена на рис. 10. Плазминоген циркулирует в крови в виде неактивного профермента и под действием различных активаторов превращается в активную сывороточную протеазу, плазмин. Существует несколько механизмов активации плазминогена. Сосудистый активатор, который выделяется клетками сосудистого эндотелия в кровоток, является основным физиологическим активатором; его выделение увеличивается под влиянием различных стимулов, в том числе закупорки венозных сосудов, действия вазоактивных веществ, физической нагрузки и гиперпирексии. Тканевый фактор, которым особенно богаты ткани матки и предстательной железы, в норме в кровоток не поступает, однако выделяется локально после обширной травмы и внесосудистого отложения фибрина. Клетки почечной паренхимы синтезируют и выделяют мочевой активатор, урокиназу, которая постоянно экскретируется с мочой и, вероятно, необходима для поддержания проходимости мочевыводящих путей. К числу других активирующих систем относится механизм, опосредованный фактором XII. Этот механизм включается вслед за активацией внутренней системы свертывания и взаимосвязан с одновременной активацией систем калликреин-кинин и комплемента [Stormorken, 1977]. Плазменные активаторы, содержание которых соответствует физиологическим уровням, быстро инактивируются антиактиваторами, а также распадаются в печени. Только в месте тромбообразования плазминоген превращается в плазмин в ощутимых количествах. Плазмин, попавший в общий кровоток, быстро инактивируется путем образования комплекса с антиплазминами [Collen, 1980]. Плазмин вызывает гидролиз фибрина до растворимых продуктов расщепления и приводит к постепенному растворению тромба. Для предотвращения неконтролируемой активности в крови большого количества взаимосвязанных реакций, приводящих к образованию фибрина, существует мощная естественная система ингибиторов свертывания. Главным ингибитором тромбина является антитромбин III, который также ингибирует активированные факторы IXa, Xa, XIa, калликреин и плазмин [Seegers, 1978]. Гепарин присоединяется к антитромбину III и изменяет его молекулярную конфигурацию, благодаря чему резко усиливается ингибиция тромбина и фактора Xa [Rosenberg, 1978]. Другими важнейшими ингибиторами тромбина плазмы являются a2-макроглобулин и a2-антитрипсин. Недавно описанный витамин К-зависимый белок, протеин С, инактивирует факторы V и VIII и таким образом, возможно, также предотвращает избыточное образование тромбина [Esmon, 1983]. По-видимому, активность обоих механизмов свертывания ограничивается не только этими ингибиторами, но и в определенной мере аутоконтролируется благодаря существованию систем обратной связи. Тромбоциты не прилипают к неповрежденным эндотелиальным клеткам, а в последнее десятилетие показано, что эндотелиальные клетки играют важную роль в ^контроле процесса гемостаза [Thorgeirsson, Robertson, 1978]. Клетки сосудистого эндотелия синтезируют из арахидоновой кислоты простациклин, который при выделении в кровь вызывает местное расширение сосудов, а также является самым мощным ингибитором адгезии и агрегации тромбоцитов [Whittle, Moncada, 1983]. Простациклин, вероятно, не циркулирует в биологически активных количествах, а выделяется местно при напряжении сосудистой стенки или ее повреждении и служит средством контроля избыточной активации тромбоцитов. Связываясь со специфическими рецепторами на мембране тромбоцита, он активирует мембранную аденилатциклазу, благодаря чему возрастает количество цАМФ. Последний ингибирует агрегацию тромбоцитов, подавляя метаболизм арахидоновой кислоты и приток кальция в клетки. Эндотелиальные клетки также синтезируют активатор плазминогена, гепариноподобный антикоагулянт, и имеют индуцируемый тромбином рецептор для протеина С. Все эти механизмы контролируют взаимодействие между кровью и стенкой сосудов, способствуя сохранению целости и проходимости сосудистого русла.
Дата добавления: 2015-11-25 | Просмотры: 721 | Нарушение авторских прав |