|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
ГЛАВА 16. ДЕГЕНЕРАТИВНЫЕ ЗАБОЛЕВАНИЯ НЕРВНОЙ СИСТЕМЫ16.1. Болезнь Паркинсона и паркинсонизм Синдром паркинсонизма проявляется замедленностью движений, мышечной ригидностью и тремором покоя. Выделяют идиопатический паркинсонизм (болезнь Паркинсона) и синдром паркинсонизма, обусловленный различными причинами и нередко служащий проявлением иных дегенеративных заболеваний нервной системы. Заболевание встречается у 60-140 на 100 000 населения; его частота резко увеличивается с возрастом. Согласно статистическим данным, паркинсонизм встречается у 1% населения до 60 лет и у 5% лиц более старшего возраста. Мужчины болеют несколько чаще, чем женщины. Этиология и патогенез. В основе заболевания лежат уменьшение количества нейронов черной субстанции и формирование в них включений - телец Леви. Его развитию способствуют наследственная предрасположенность, пожилой и старческий возраст, воздействие экзогенных факторов. В возникновении акинетико-ригидного синдрома могут иметь значение наследственно обусловленное нарушение обмена катехоламинов в мозге или неполноценность ферментных систем, контролирующих этот обмен. Часто выявляется семейная отягощенность по этому заболеванию при аутосомно-доминантном типе наследования. Синдром паркинсонизма возникает в результате перенесенных острых и хронических инфекций нервной системы (клещевой и другие виды энцефалитов). Причинами болезни могут служить острые и хронические расстройства мозгового кровообращения, травмы и опухоли нервной системы. Возможно развитие паркинсонизма вследствие лекарственных интоксикаций при длительном использовании препаратов фенотиазинового ряда (аминазин, трифтазин), метилдофы, некоторых наркотических средств. Паркинсонизм может развиваться при острой или хронической интоксикации окисью углерода и марганца. Воздействие различных экзо- и эндогенных факторов способствует проявлению генуинных дефектов в механизмах обмена катехоламинов в подкорковых ядрах и возникновению заболевания. Синдром паркинсонизма может быть одним из проявлений поражения ЦНС при нейро- дегенеративных заболеваниях (кортикобазальная дегенерация, множественные системные атрофии, болезнь Вильсона-Коновалова и пр.). Ведущим патогенетическим звеном как болезни Паркинсона, так и синдрома паркинсонизма является нарушение обмена дофамина в экстрапирамидной системе. Дофамин выполняет медиаторную функцию в реализации двигательных актов, основное место его синтеза - черная субстанция. Аксоны дофаминергических нейронов, расположенные в черной субстанции (нигростриарные нейроны), направляются к стриатуму. Воздействуя через дофаминергические рецепторы различных типов, они уменьшают выраженность ингибирующего влияния базальных ганглиев, облегчают таламокортикальную передачу, приводя тем самым к «растормаживанию» моторной коры. В норме эффективное функционирование экстрапирамидной системы достигается балансом между дофаминергической и холинергической системами черной субстанции и хвостатого ядра, а также дофаминергической и ГАМКергической системами черной субстанции и стриатума, функционирующими по принципу обратной связи. При нарушении функции черной субстанции наступает блокада импульсов, поступающих из экстрапирамидных зон коры большого мозга и полосатого тела к передним рогам спинного мозга. В то же время к клеткам передних рогов поступают ингибирующие импульсы из бледного шара и черной субстанции. В результате усиливается циркуляция импульсов в системе альфа- и гаммамотонейронов спинного мозга с преобладанием альфа-активности, что приводит к возникновению экстрапирамидной ригидности. В патогенезе также имеет значение нарушение деятельности мозговых структур, использующих в качестве нейротрансмиттеров норадреналин, субстанцию Р, глутамат. Патоморфология. Основные патологоанатомические изменения при паркинсонизме наблюдаются в черном веществе и бледном шаре в виде дегенеративных изменений и гибели нервных клеток. На месте погибших клеток возникают очаги разрастания глиальных элементов или остаются пустоты. В поздних стадиях заболевания выявляется атрофия коры больших полушарий. Кроме того, в дофамин- и норадренергических нейронах, а также в клетках коры больших полушарий телец Леви определяются включения, содержащие сфингомиелин. Клинические проявления. Основой клинических проявлений является акинетико-ригидный или гипертонически-гипокинетический синдром, включающий в себя триаду: олиго-, брадикинезию, мышечную ригидность и тремор. Развивается своеобразная сгибательная поза: голова и туловище наклонены вперед, руки полусогнуты в локтевых, лучезапястных и фаланговых суставах, нередко плотно приведены к боковым поверхностям грудной клетки, ноги полусогнуты в коленных суставах. Резко обеднена гамма двигательных синкинезий. Мимика также бедная (гипоили амимия). Речь тихая, монотонная, без модуляций, с наклонностью к затуханию в конце фразы. Темп произвольных движений с развитием заболевания постепенно замедляется вплоть до полной обездвиженности. Больной ходит мелкими шаркающими шагами. При ходьбе отсутствуют содружественные движения рук (ахейрокинез). Развивается постуральная неустойчивость - ограничение способности поддерживать положение центра тяжести, особенно при движении. Нередко наблюдается склонность к непроизвольному бегу вперед (пропульсии). Если слегка толкнуть больного вперед, он бежит, чтобы не упасть, как бы «догоняя свой центр тяжести». Аналогично легкий толчок в грудь ведет к тому, что больной делает несколько шагов назад (ретропульсии), в сторону (латеропульсии). Эти движения наблюдаются также при попытке сесть, встать, откинуть голову назад. При исследовании мышечного тонуса в конечностях отмечаются своеобразное мышечное сопротивление вследствие повышения тонуса мышц-антагонистов, феномен «зубчатого колеса» (создается впечатление, что суставная поверхность состоит из сцепления двух зубчатых колес). Равномерное повышение тонуса в мышцах-антагонистах (сгибатели-разгибатели, пронаторы-супинаторы) носит название пластического мышечного тонуса. Повышение тонуса при пассивных движениях можно определить следующим приемом: если поднять голову лежащего пациента, а потом резко отпустить руку, то голова не упадет на подушку, а опустится относительно плавно. Иногда голова лежащего больного несколько приподнята (феномен «воображаемой подушки»). Тремор - характерный, хотя и не обязательный для синдрома паркинсонизма, симптом. Это ритмичное, регулярное, непроизвольное дрожание конечностей, лицевой мускулатуры, головы, нижней челюсти, языка, более выраженное в покое и уменьшающееся при активных движениях. Частота колебаний 4-6 в секунду. Иногда отмечаются движения пальцами в виде «скатывания пилюль», «счета монет». Тремор усиливается при волнениях и практически исчезает во сне. Психические нарушения проявляются утратой инициативы, снижением активности, сужением круга интересов, резким угнетением различных эмоциональных реакций и аффектов, а также некоторой поверхностностью и медлительностью мышления (брадифрения). Наблюдаются брадипсихия - затруднение активного переключения с одной мысли на другую, акайрия - прилипчивость, вязкость, эгоцентризм. По мере прогрессирования заболевания у значительного числа больных развиваются когнитивные нарушения, нередко достигающие степени деменции. Эмоциональные нарушения представлены депрессией. Вегетативные нарушения проявляются в виде сальности кожи лица и волосистой части головы, себореи, гиперсаливации, гипергидроза, трофических нарушений в дистальных отделах конечностей. Иногда специальными методами исследования определяется нерегулярное по частоте и глубине дыхание. В зависимости от преобладания тех или иных симптомов выделяют ригидно-брадикинетическую, дрожательно-ригидную и дрожательную формы заболевания. Ригидно-брадикинетическая форма проявляется повышением тонуса мышц по пластическому типу, прогрессирующим замедлением активных движений вплоть до полной обездвиженности, флексорной (сгибательной) позой больных. Эта форма паркинсонизма, наиболее неблагоприятная по течению, чаще наблюдается при сосудистом и реже - при постэнцефалитическом паркинсонизме. Дрожательно-ригидная форма включает в себя тремор конечностей, преимущественно их дистальных отделов, к которому с развитием заболевания присоединяется скованность произвольных движений. При дрожательной форме паркинсонизма отмечается постоянный или почти постоянный средне- и крупноамплитудный тремор конечностей, языка, головы, нижней челюсти. Тонус мышц нормальный или несколько повышен по пластическому типу. Темп произвольных движений сохранен. Эта форма чаще встречается при постэнцефалитическом и посттравматическом паркинсонизме. Если синдром паркинсонизма служит одним из проявлений поражения нервной системы, можно определить и другие неврологические нарушения. При сосудистом и постэнцефалитическом паркинсонизме могут выявляться признаки пирамидной недостаточности, псевдобульбарный синдром. При постэнцефалитическом паркинсонизме встречаются так называемые окулогирные кризы - фиксация взора кверху в течение нескольких минут или часов; иногда голова при этом запрокинута. Кризы могут сочетаться с нарушением конвергенции и аккомодации (прогрессирующий надъядерный паралич). Течение болезни Паркинсона прогрессирующее. В начале заболевания могут преобладать симптомы на одной стороне, с течением времени симптоматика становится двусторонней. Течение синдрома паркин- сонизма обусловлено причиной основного заболевания. Так, при некоторых формах, вызванных лекарственными интоксикациями, при отмене препаратов может наступить улучшение состояния. Диагностика и дифференциальная диагностика. В первую очередь следует дифференцировать болезнь Паркинсона от синдрома паркинсонизма. При постэнцефалитическом паркинсонизме есть острое инфекционное поражение ЦНС в анамнезе. При осмотре выявляются глазодвигательные расстройства; могут наблюдаться спастическая кривошея, торсионная дистония, которые никогда не выявляются при болезни Паркинсона. Посттравматический паркинсонизм возникает как следствие тяжелой черепно-мозговой травмы, иногда повторной. Часто встречаются вестибулярные расстройства, нарушение интеллекта и памяти, очаговые симптомы вследствие поражения вещества головного мозга. Для диагностики токсического паркинсонизма имеют значение анамнез (сведения о работе в контакте с марганцем или его окислами, прием нейролептиков, контакт с другими токсинами), обнаружение их метаболитов в биологических жидкостях. При сосудистом паркинсонизме дрожание и ригидность сочетаются с другими признаками сосудистого поражения мозга или возникают после острых нарушений мозгового кровообращения. Выявляются очаговые неврологические симптомы в виде пирамидной недостаточности, выраженные псевдобульбарные симптомы. Имеются инструментальные признаки сосудистого заболевания, по данным нейровизуализации выявляются очаговые поражения вещества мозга. Инструментальных и лабораторных признаков, специфичных для болезни Паркинсона, нет. При изучении мозгового кровотока методом однофотонной эмиссионной КТ могут определяться зоны сниженного кровотока в базальных ганглиях. Результаты позитронно-эмиссионной томографии позволяют установить снижение метаболизма в стриатуме. Лечение. Своевременно начатое лечение позволяет уменьшить выраженность симптомов и замедлить прогрессирование заболевания. В поздних стадиях лечебные мероприятия менее эффективны. В ранних стадиях заболевания применяют агонисты дофаминовых рецепторов (бромокриптин, пирибедил, прамипексол), амантадин, селективные ингибиторы моноаминоксидазы В (селегилин), ингибиторы катехол-О-метилтрансферазы (толкапон, энтокапон), антихолинэстеразные препараты (циклодол). Дозы и комбинации препаратов подбирают индивидуально с учетом индивидуальной переносимости и клинической картины. Так, назначение агонистов дофаминовых рецепторов позволяет эффективно контролировать нарушения тонуса и другие двигательные расстройства, но могут вызывать артериальную гипотензию, нарушения сна, зрительные галлюцинации. Побочные эффекты ограничивают применение центральных холинолитиков. При выраженных клинических проявлениях паркинсонизма в настоящее время средством выбора являются препараты леводопы, которая, попадая в ЦНС, декарбоксилируется в дофамин, необходимый для нормальной функции базальных ганглиев. При сочетании леводопы с ингибитором декарбоксилазы можно уменьшить дозу леводопы и тем самым уменьшить риск развития побочных явлений. С этой целью применяют комбинированные препараты, содержащие леводопу и карбидопу (наком, синемет) или леводопу и бензеразид (мадопар) в соотношении 10:1 или 4:1. Препараты влияют прежде всего на акинезию и в меньшей степени на другие симптомы. Максимального эффекта позволяет добиться многократный в течение суток прием препаратов для создания равномерной концентрации лекарственного средства в крови, а также комбинация медленно высвобождающихся и быстродействующих форм. Лечение начинают с минимальных доз. Дозы увеличивают медленно, в течение нескольких недель, до получения клинического эффекта. Побочные действия препарата - дистонические нарушения, психозы, колебания АД. Возможна комбинация препаратов леводопы и других противопаркинсонических средств. При неэффективности консервативной терапии обсуждается вопрос о целесообразности хирургического лечения, принципы которого изложены в соответствующей главе. 16.2. Хорея Гентингтона Хроническое прогрессирующее наследственно-дегенеративное заболевание с нарастающим хореическим гиперкинезом и деменцией. Частота составляет от 2 до 7 случаев на 100 000 населения. Заболевание передается аутосомно-доминантно с высокой пенетрантностью (80-85%). Молекулярной основой хореи Гентингтона является экспансия повторов CAG в гене, ответственном за синтез белка гентингтина (4р16.3). В норме имеется от 10 до 35 повторов. В мутантном гене насчитывается от 36 до 200 повторов, что сопровождается синтезом дефектного гентингтина, имеющего аномально длинную последовательность из остатков глутаминовой кислоты. При передаче гена от отца отмечается большее увеличение числа повторов, что сопровождается развитием более ранних и тяжелых форм заболевания. Гентингтин вырабатывается в головном мозге, особенно активно в коре и в мозжечке, и присутствует как в цитоплазме, так и в ядре нервных клеток. Цитоплазматический гентингтин может участвовать в транспорте везикул и в поддержании цитоскелета. В ядрах нейронов гентингтин участвует в определенных стадиях дифференцировки клеточного цикла. Считается, что при протеолизе гентингтина образуются токсичные для клеток фрагменты, содержащие полиглутамины. В норме их немного, и они утилизируются. При болезни Гентингтона добавочные CAG-повторы приводят к увеличению числа полиглутаминовых остатков и усилению их токсического влияния на клетку. Патоморфология. В подкорковых ганглиях, преимущественно в скорлупе и хвостатом ядре, определяются выраженные дегенеративные изменения мелких и крупных клеток, уменьшение их числа, разрастание глиальных элементов, отмечается расширение наружных и внутренних ликворопроводящих пространств. Клинические проявления. Заболевание возникает обычно в возрасте 30 лет и старше. Первыми симптомами могут быть интеллектуальные расстройства, в дальнейшем постепенно развивается деменция. Одновременно появляются хореические гиперкинезы: быстрые, неритмичные, беспорядочные движения в различных мышечных группах. Выполнение произвольных движений затруднено вследствие гиперкинезов и сопровождается рядом ненужных движений. Так, например, при ходьбе больные жестикулируют, приседают, широко расставляют руки (рис. 16.1). Однако даже при выраженном гиперкинезе, особенно в начале болезни, они могут его сознательно подавлять на некоторое время. Речь затруднена и также сопровождается излишними движениями, в результате гиперкинезов звуковоспроизводящей мускулатуры появляются непроизвольные вскрики, всхлипывания. Отмечаются гиперкинезы мимической мускулатуры. Мышечный тонус снижен. Парезы конечностей и другие очаговые неврологические симптомы не определяются. Нередко наблюдаются эндокринные и нейротрофические расстройства.
Психические и поведенческие нарушения обусловлены синдромом гиперактивности: дефицитом внимания, немотивированной сменой настроения, расторможенностью, снижением критики своего состояния. В отдельных случаях наблюдаются бредовые расстройства, деменция. В 5-16% случаев диагностируют атипичный акинетико-ригидный вариант хореи Гентингтона. При этом развивается акинетико-ригидный синдром в сочетании с прогрессирующей интеллектуальной деградацией и умеренно выраженным хореическим гиперкинезом. Из насильственных движений преобладает хореоатетоз. Заболевание неуклонно прогрессирует. Его длительность 5-10 лет с момента возникновения первых симптомов. Более доброкачественное течение отмечается при атипичной акинетико-ригидной форме. Диагностика и дифференциальная диагностика. Во всех случаях большое значение имеют отягощенность семейного анамнеза по этому заболеванию и результаты молекулярно-генетического анализа. Диагностика может вызывать затруднения в атипичных случаях хореи Гентингтона. Результаты инструментального обследования неспецифичны: на ЭЭГ отмечаются диффузные изменения биоэлектрической активности мозга. При КТ и МРТ выявляются признаки атрофии мозгового вещества - расширение желудочков и субарахноидальных пространств. Дифференцировать хорею Гентингтона следует от малой хореи, хореического гиперкинеза при очаговых поражениях головного мозга (опухоль, инсульт, энцефалит), а также от старческой (сенильной) хореи. Лечение. Для подавления гиперкинезов и купирования эмоциональных нарушений назначают антагонисты дофамина. Используют нейролептики: галоперидол, пимозид, оланзапин; дозы подбирают индивидуально. При атипичных формах с преобладанием акинетико-ригидных расстройств применяют агонисты дофаминовых рецепторов. 16.3. Торсионная дистония Генетически гетерогенная группа заболеваний, клинически проявляющаяся изменениями мышечного тонуса и непроизвольными тоническими сокращениями мышц туловища и конечностей. Различают идиопатическую (семейную) торсионную и симптоматическую дистонию. Тип наследования идиопатической торсионной дистонии как аутосомно-доминантный, так и аутосомно-рецессивный. Развитие семейной формы торсионной дистонии связано с несколькими мутациями, из которых чаще встречается аутосомно-доминантная форма, обусловленная делецией тринуклеотидного повтора GAC в гене, локализованном на хромосоме 9 (9q34) и ответственном за выработку торзина А. Симптоматическая торсионная дистония встречается при гепатоцеребральной дистрофии, хорее Гентингтона, опухолях мозга, эпидемическом энцефалите, детском церебральном параличе. Патоморфология. Дистрофические изменения обнаруживаются преимущественно в мелких нейронах в области скорлупы чечевицеобразного ядра, реже - в других базальных ганглиях. Клинические проявления. Заболевание развивается постепенно, чаще в возрасте до 15 лет. В детском возрасте первыми симптомами болезни могут быть нарушение походки, спастическая кривошея; у взрослых чаще встречаются первичные генерализованные формы. В результате нарушения соотношения функций мышц-синергистов и антагонистов возникают насильственные длительные тонические сокращения мышц туловища, головы, тазового пояса, конечностей, обычно ротаторные, сочетающиеся с атетоидными движениями в пальцах. Создается впечатление, что мышцы постоянно сокращаются для преодоления действия антагонистов. Возникающие позы, даже самые неудобные, долго сохраняются. Гиперкинезы усиливаются при волнении, активных движениях, во сне исчезают. По мере прогрессирования заболевания дистонии становятся постоянными, усиливается поясничный лордоз. В зависимости от распространенности дистонических явлений выделяют локальную и генерализованную формы заболевания. При локальных дистонических симптомах возникает тоническое сокращение отдельных мышечных групп, нарушаются произвольные движения, возникает аномальная поза. К таким симптомам относятся спастическая кривошея, писчий спазм, оромандибулярная дистония (открывание и закрывание рта, непроизвольные движения языка), блефароспазм, щечно-лицевая, щечно-язычная дистония, хореоатетоз. Заболевание в большинстве случаев неуклонно прогрессирует, но иногда отмечаются ремиссии различной длительности. Диагностика и дифференциальная диагностика. Большое число спорадических и симптоматических форм дистоний делает диагностику достаточно сложной. Необходимо исключить заболевания, на фоне которых развились гиперкинезы (воспалительные, сосудистые и прочие поражения головного мозга). Лечение симптоматическое, применяют нейролептики (галоперидол, пимозид), холинолитики (циклодол). Положительный эффект мо- гут дать антиконвульсанты (карбамазепин, клоназепам). В случаях с преобладанием мышечной ригидности эффективно использование препаратов леводопы (наком, мадопар, синемет). При локальных формах дистонии (спастическая кривошея, оромандибулярная дискинезия) возможно локальное введение препаратов ботулотоксина. В ряде случаев проводятся стереотаксические операции с деструкцией вентролатерального ядра таламуса и субталамической области. 16.4. Гепатоцеребральная дистрофия Гепатоцеребральная дистрофия (гепатолентикулярная дегенерация, болезнь Вестфаля-Вильсона-Коновалова) - хроническое прогрессирующее наследственно-дегенеративное заболевание с сочетанным поражением подкорковых узлов ЦНС и печени. Частота составляет 2-3 случая на 100 000 населения. Передается аутосомно-рецессивно. Ген картирован на длинном плече хромосомы 13 (13q 14.3). Белковым продуктом гена является АТФаза, участвующая в синтезе церулоплазмина и элиминации меди из тканей. Нарушение синтеза церулоплазмина сопровождается нарушением транспорта меди, вследствие чего происходит ее отложение в органах и тканях, преимущественно в печени, мозге, роговице, а также в почках и других органах. Избыточное отложение меди приводит к блоку сульфгидрильных групп в окислительных ферментах и к нарушению окислительно-восстановительных процессов в клетке. Патоморфология. В мозге, печени, почках, селезенке, роговице, радужной оболочке, хрусталике глаза определяются дегенеративные изменения, наиболее выраженные в подкорковых ядрах. Обнаруживаются также дистрофические изменения нервных клеток, очаговые размягчения мозговой ткани с образованием кист, разрастанием глии. Выявляются изменения мелких сосудов мозговой ткани, кровоизлияния вокруг них, периваскулярный отек. Постоянным признаком, особенно при длительном течении заболевания, является цирроз печени. Клинические проявления складываются из симптомов поражения ЦНС и внутренних органов. У больных появляются и нарастают мышечная ригидность, разнообразные гиперкинезы, псевдобульбарные симптомы, прогрессирующее снижение интеллекта, нарушения функции печени и изменение радужной оболочки (кольцо Кайзера-Флей- шера). Ведущим является синдром экстрапирамидных расстройств: ригидность мышц туловища, конечностей, глотки и, как следствие этого, нарушения походки, глотания, речи. Параллельно возникают различные гиперкинезы: тремор, атетоз, торсионная дистония, интенционное дрожание, усиливающиеся при попытке выполнения произвольных движений. У большинства больных имеется прогрессирующее нарушение функции печени, значительно осложняющее прогноз заболевания. В зависимости от выраженности и сочетания клинических проявлений, возраста, в котором возникло заболевание, и степени поражения печени выделяют 5 форм гепатоцеребральной дистрофии. Абдоминальная форма сопровождается преимущественным нарушением функции печени. Ранняя ригидно-аритмогиперкинетическая форма имеет наиболее злокачественное течение. Неврологические проявления развиваются в возрасте 7-15 лет. Этому, как правило, предшествуют признаки поражения печени. В клинической картине преобладают мышечная ригидность и гиперкинезы. Дрожательно-ригидная и дрожательная формы, проявляющиеся в более позднем возрасте (17-20 лет), сопровождаются одновременно ригидностью и дрожанием, что часто бывает первым признаком заболевания. Дрожание, постепенно усиливаясь, может становиться генерализованным и вовлекать мышцы туловища, конечностей, лица, голосовых связок, дыхательную мускулатуру, диафрагму. Нарушается глотание, речь становится скандированной. Часто отмечаются выраженные изменения психики. Экстрапирамидно-корковая форма отличается расстройством высших мозговых функций, параличами, часто эпилептическими припадками, грубым снижением интеллекта с изменением личности. Течение неуклонно прогрессирующее. Продолжительность жизни зависит от клинической формы заболевания и своевременности начатого лечения. Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании клинических симптомов и данных лабораторных методов обследования. Патогномоничным признаком гепатоцеребральной дистрофии является роговичное кольцо Кайзера-Флейшера, обусловленное отложением пигмента, содержащего медь, по периферии радужной оболочки. В сыворотке крови значительно снижено содержание церулоплазмина (ниже 10 ЕД при норме 25-45 ЕД), отмечаются гипопротеинемия, гиперкупрурия (до 1000 мкг/сут и выше при норме 150 мкг/сут) и гипераминоацидурия (до 1000 мг/сут при норме 350 мг/сут). Возможны также повышение содержания аммиака в крови, изменение печеночных проб. Соответствующий семейный анамнез, клиническая картина, роговичное кольцо Кайзера-Флейшера, низкий уровень церулоплазмина в крови и повышение экскреции меди с мочой у больных и их родственников позволяют диагностировать гепатоцеребральную дистрофию. Диагноз подтверждается результатами молекулярно-генетического исследования. Заболевание следует дифференцировать от малой хореи, дегенеративных подкорковых заболеваний, рассеянного склероза, а также от поражений нервной системы на фоне хронической печеночной недостаточности. Лечение. Основной целью лечения является выведение из организма избытка меди. Для этого используют тиоловые препараты, к которым относятся унитиол, декаптол и D-пеницилламин. Дозы подбирают индивидуально. D-пеницилламин назначают в средних дозах от 0,45 до 2 г/сут после еды. Препарат необходимо принимать в течение всей жизни. Наиболее эффективно лечение в ранних стадиях болезни. Унитиол назначают повторными курсами по 5 мл 5% раствора внутримышечно ежедневно или через день (на курс 25 инъекций с перерывом между курсами 5-6 мес). Используют препараты цинка, препятствующие всасыванию меди в кишечнике, что позволяет снизить дозу D-пенициллами- на, а также средства, улучшающие функции печени. В качестве симптоматических средств для купирования гиперкинезов возможно применение антиконвульсантов, нейролептиков (следует использовать низкие дозы препаратов, учитывая нарушение функций печени). Рекомендуется специальная диета с ограничением продуктов, богатых медью (печень, грибы, шоколад, устрицы и др.), животных жиров, белков. Пища должна быть богата витаминами и углеводами. 16.5. Семейная спастическая параплегия (болезнь Штрюмпеля) Семейная спастическая параплегия - гетерогенная группа заболеваний с двусторонним поражением пирамидных путей в боковых и передних канатиках спинного мозга. Выделяют формы с изолированным поражением пирамидных путей и с вовлечением других отделов нервной системы. К настоящему времени на основании молекулярно-генетического дефекта выделено 8 вариантов заболевания, передающихся аутосомно-доминантно, аутосомно-рецессивно или сцепленно с Х-хромосомой. Для отдельных форм установлены мутации и определены конечные продукты экспрессии гена. Наиболее частая форма изолирован- ной спастической параплегии у взрослых (около 50-60% случаев) наследуется по аутосомно-доминантному типу с локализацией мутации на длинном плече хромосомы 14 (14q11.2-24.3). Дефект включает точечную мутацию, приводящую к нарушению синтеза спастина, - белка, принимающего участие в регуляции мышечного тонуса. Реже изолированная спастическая параплегия взрослых может развиваться при мутациях на хромосомах 15 и 16 (15q11.1 и 16q24.3 соответственно). Мутация на хромосоме 15 связана с нарушением синтеза белка параплегина, относящегося к классу митохондриальных белков - АТФаз. Мутации на Х-хромосоме (Xq21-22) в гене, ответственном за белок миелиновый протеолипид, сопровождаются выраженным клиническим разнообразием, начиная от тяжело протекающих форм раннего детского возраста (болезнь Пелицеуса-Мерцбахера) до относительно мягких форм изолированной спастической параплегии взрослых. Патоморфология. Наиболее часто поражаются поясничная и грудная части спинного мозга, реже ствол головного мозга. Отмечается симметричное глиозное перерождение пирамидных путей в боковых и передних канатиках. В меньшей степени вовлекаются задние канатики. Описаны случаи дегенеративных изменений в клетках коры передней центральной извилины, передних рогов спинного мозга, мозжечковых проводниках. Клинические проявления. Развитие заболевания постепенное, первые симптомы появляются на втором десятилетии жизни, хотя отмечаются колебания возраста дебюта заболевания. Первые проявления - скованность в ногах и повышенная утомляемость при ходьбе, нарастающие по мере прогрессирования заболевания. Постепенно развивается спастическая походка, присоединяются варусная и эквиноварусная деформации стоп, изменения стоп по типу стопы Фридрейха, контрактуры, особенно выраженные в голеностопных суставах. Слабость в нижних конечностях с течением времени нарастает, но полного паралича нижних конечностей не наблюдается. При клиническом обследовании больных уже в начальных стадиях заболевания обнаруживается повышение сухожильных рефлексов, рано появляются патологические рефлексы сгибательной и разгибательной групп, клонусы стоп, надколенников. Значительно позже в патологический процесс вовлекаются верхние конечности. У отдельных больных могут наблюдаться поражение зрительных и глазодвигательных нервов, нистагм, дизартрия, атаксия и интенционный тремор. Кожные рефлексы в большинстве случаев сохраняются, функции тазовых органов не нарушены. Расстройства чувствительности отсутствуют, интеллект сохранен. При аутосомно-доминантном типе наследования выделяют два варианта относительно благоприятного течения. При первом варианте, встречающемся чаще, заболевание начинается в возрасте до 35 лет, медленно прогрессирует, выраженных парезов нет. При втором варианте симптомы появляются обычно в более позднем возрасте (после 35-40 лет), но быстро нарастают с развитием выраженной спастичности, парезов и более тяжелой инвалидизацией больных. Чаще вовлекаются другие отделы нервной системы: в частности, отмечаются расстройства глубокой чувствительности и тазовых функций. Течение заболевания медленно прогрессирующее; прогноз для жизни благоприятный. Диагностика и дифференциальная диагностика. Диагностика обычно не вызывает затруднений при семейных случаях заболевания и типичной клинической картине. На МРТ спинного мозга выявляются атрофические изменения в боковых и передних канатиках и в меньшей степени в задних столбах. Дифференциальная диагностика проводится с рассеянным склерозом, боковым амиотрофическим склерозом, опухолями спинного мозга и другими патологическими процессами, вызывающими компрессию спинного мозга, мозжечково-пирамидными дегенерациями. Лечение симтоматическое - миорелаксанты лиорезал (баклофен), тизанидин (сирдалуд), толперизон (мидокалм). Целесообразно проведение курсового общеукрепляющего лечения, включающего в себя витамины группы В, метаболические препараты: пирацетам (ноотропил), пиридитол (энцефабол), церебролизин, аминокислоты. Показаны физиотерапевтические процедуры: парафиновые аппликации на мышцы нижних конечностей, массаж, рефлексотерапия, ЛФК, при необходимости ортопедические мероприятия. 16.6. Спиноцеребеллярные атаксии Аутосомно-рецессивные атаксии Аутосомно-рецессивные атаксии включают несколько заболеваний, из которых наиболее частым является семейная атаксия Фридрейха. Семейная атаксия Фридрейха - наследственное дегенеративное заболевание нервной системы с поражением задних и боковых канатиков спинного мозга. Тип наследования - аутосомно-рецессивный с неполной пенетрантностью патологического гена. Распространенность составляет 2-5 случаев на 100 000 населения. Патогенез и патоморфология. Ген, ответственный за заболевание, локализован на хромосоме 9 (9q13-21.1) и кодирует синтез митохондриального белка фратаксина, участвующего в транспорте железа в митохондриях. Мутация в большинстве случаев заключается в экспансии тринуклеотидных повторов GAA (гуанин-аденин-аденин) на обоих аллелях в гене фратаксина, реже возможно сочетание экспансии триплетов с аллельной точечной мутацией. В норме на этом участке имеется 10-25 повторов, в то время как при болезни Фридрейха число триплетов увеличено в 3-10 раз и более (от 100 до 2000 и более). Экспансия повторов приводит к нарушению синтеза фратаксина, что сопровождается накоплением ионов железа в митохондриях, усилением свободнорадикальных реакций с деструкцией митохондриальных мембран и развитием нарушений аэробного дыхания в клетках и тканях. В первую очередь поражаются наиболее энергозависимые органы: ЦНС, сердце, скелетная мускулатура, эндокринные органы. В патологический процесс вовлекаются задние и боковые канатики спинного мозга (пути Голля и Бурдаха, Флексига и Говерса, пирамидные тракты), а также чувствительные спинномозговые ганглии и чувствительные волокна периферических нервов. Изменения в мозжечке, стволе и других отделах головного мозга отмечаются в более поздней стадии болезни. Клинические проявления. Начало заболевания относится к 6-15-лет- нему возрасту. Первым симптомом болезни является неустойчивая походка, которую Шарко назвал табетически-мозжечковой. В ранних стадиях атаксия выражена преимущественно в ногах и имеет заднестолбовое происхождение. По мере прогрессирования заболевания нарушения координации распространяются на верхние конечности и атаксия может стать сочетанной. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Нарушается выполнение тонких движений, меняется почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов; мышечный тонус снижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередки патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен. Выявляются симптомы экстраневрального поражения: изменения сердца (кардиомиопатия), костей (сколиоз, укороченная стопа с высоким сводом - стопа Фридрейха), зрения (атрофия зрительных нервов), эндокринной системы (диабет, гипогонадизм).
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании деформаций стоп (рис. 16.2) по типу стопы Фридрейха, поражения миокарда, эндокринных расстройств и молекулярно-генетического анализа. Используется генетическая диагностика. Атаксию Фридрейха необходимо дифференцировать от атаксии, обусловленной дефицитом витамина Е (синдром AVED - ataxia and vitamin E deficiency). Заболевание передается аутосомно-рецессивно и связано с точечной мутацией на хромосоме 8 (8q13.1-13.3) в гене, кодирующем белок-переносчик α-токоферола. В результате мутации содержание α-токоферола в плазме снижается, что приводит к нарушению антиоксидантной функции α-токоферола в митохондриальных мембранах. В этих случаях постоянный прием витамина Е в больших дозах приводит к полному регрессу неврологических симптомов. Атаксию Фридрейха следует отличать также от других форм мозжечковых дегенераций, фуникулярого миелоза, рассеянного склероза. Лечение. При атаксии Фридрейха применяют симптоматические средства, общеукрепляющие препараты, ЛФК, массаж. Положительный эффект могут давать препараты, оказывающие воздействие на метаболизм нервной ткани (церебролизин, ноотропил, карнитин). Аутосомно-доминантные спиноцеребеллярные атаксии (наследственные спиноцеребеллярные атаксии) К настоящему времени установлено более 13 молекулярно-генетических дефектов, приводящих к развитию этой группы заболеваний. Основная классификация формы заболевания проводится на основании генетического дефекта. В Российской Федерации наиболее частой формой является спиноцеребеллярная атаксия (СЦА) 1-го типа. Патогенез. Аутосомно-доминантные атаксии (наследственные спиномозжечковые атаксии) - группа заболеваний, развивающихся вследствие экспансии тандемных тринуклеотидных повторов CAG (цито- зин-аденин-гуанин). В норме число повторов составляет 15-25, а при болезни оно увеличено до 40 и более. Кодон CAG кодирует глутамин. Увеличение числа повторов приводит к удлинению полиглутаминовых цепей в составе белка, что сопровождается возникновением нерастворимых связей и, как следствие, этого накоплением белковых включений и гибелью клетки. Имеется прямая связь между числом тринуклеотидных повторов, возрастом развития заболевания и тяжестью болезни. Клинические проявления. Все формы сопровождаются клиникой мозжечкового поражения в виде статической и динамической атаксии, нарушения походки, скандированной речи, интенционного дрожания, нистагма и других симптомов. Характер и выраженность неврологического дефицита, сроки дебюта заболевания и тяжесть его течения определяются особенностями генетического дефекта и связанными с ним изменениями экспрессируемого белка. Так, при СЦА 1-го типа, помимо мозжечковых нарушений, в клинической картине представлены пирамидные расстройства, а при СЦА 3-го типа (болезнь Мачадо-Джозефа), кроме того, выявляются наружная офтальмоплегия, амиотрофия, моторно-сенсорная полинейропатия. СЦА 1-4 типа начинаются на 3-4 десятилетиях жизни, продолжительность жизни при них составляет 10-20 лет, тогда как СЦА 5-6 типа начинаются в возрасте 45-55 лет, проявляются изолированной атаксией ходьбы и имеют благоприятное течение. СЦА 7-го типа сопровождается прогрессирующей атрофией сетчатки.
Лечение симптоматическое. Определенный эффект могут давать препараты, способствующие нормализации метаболизма нервной ткани, антиоксиданты. Спорадические спиноцеребеллярные атаксии Оливопонтоцеребеллярная атаксия Дежерина-Тома является одной из форм мультисистемных дегенераций, при которых в патологический процесс вовлекаются различные структуры ЦНС. Морфологически проявляется дегенерацией коры больших полушарий, червя мозжечка, нижних олив, черной субстанции и других базальных ганглиев, таламуса, клеток передних и боковых рогов спинного мозга. Заболевание начинается на 4-5 десятилетии жизни с появления атактической походки, впоследствии атаксия становится генерализованной, присоединяются симптомы поражения других отделов нервной системы: бульбарные расстройства, тазовые нарушения. Продолжительность заболевания составляет 10-15 лет. Поздняя корковая мозжечковая атрофия Мари-Фуа-Алажуанина. Заболевают преимущественно мужчины, начало заболевания - в возрасте после 50 лет. В клинической картине преобладает динамическая и туловищная атаксия. Менее выражены бульбарные и вестибулярные нарушения. При использовании методов нейровизуализации у части больных выявляется атрофия червя и полушарий мозжечка. Дифференциальная диагностика проводится с аутосомно-доминантными формами спиноцеребеллярных атаксий, мультисистемными дегенерациями, рассеянным склерозом. Дата добавления: 2015-12-16 | Просмотры: 1575 | Нарушение авторских прав |
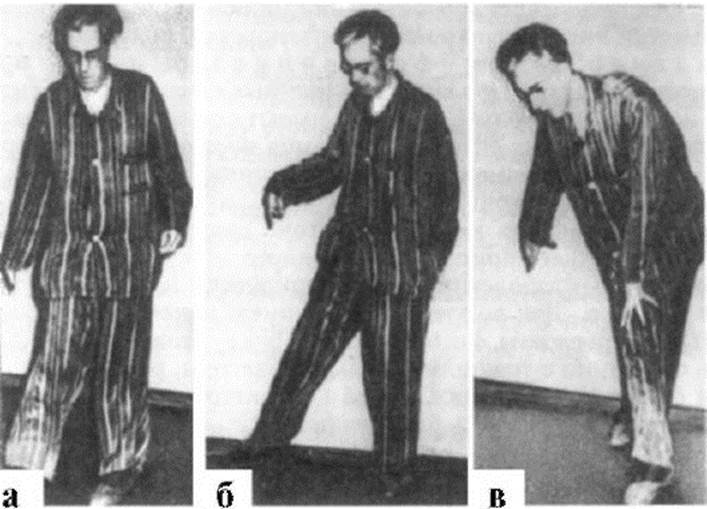 Рис. 16.1. Хорея Гентингтона (а-в)
Рис. 16.1. Хорея Гентингтона (а-в)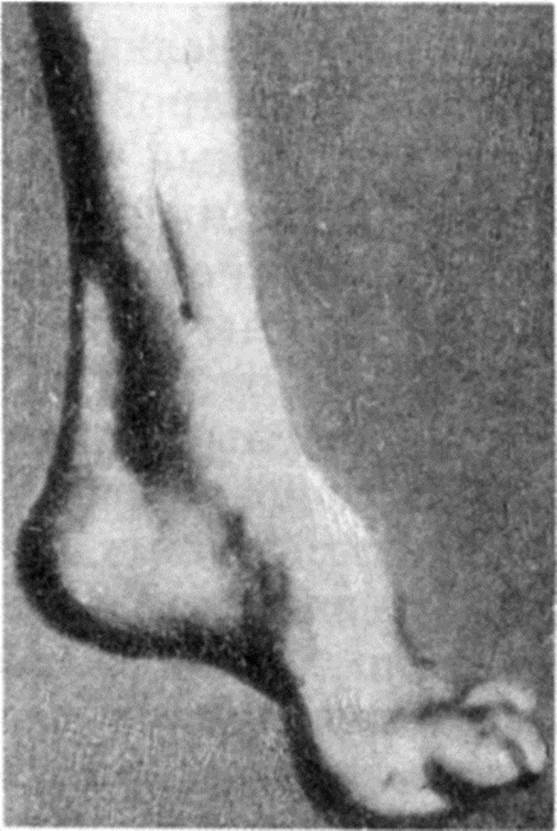 Рис. 16.2. Стопа Фридрейха
Рис. 16.2. Стопа Фридрейха Диагностика и дифференциальная диагностика. Диагноз заболевания устанавливается на основании особенностей клинической картины и результатов молекулярно-генетического анализа. Дифференциальная диагностика проводится с атаксией Фридрейха, рассеянным склерозом, дегенеративными заболеваниями с преимущественным поражением мозжечка.
Диагностика и дифференциальная диагностика. Диагноз заболевания устанавливается на основании особенностей клинической картины и результатов молекулярно-генетического анализа. Дифференциальная диагностика проводится с атаксией Фридрейха, рассеянным склерозом, дегенеративными заболеваниями с преимущественным поражением мозжечка.