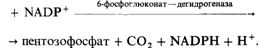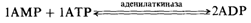|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Типичные нарушения функций ферментов: ферменты эритроцитовК настоящему времени подробно изучена группа наследственных заболеваний, связанных с недостаточностью ферментативных систем эритроцитов [933, 1345]. Эритроциты человека – безъядерные клетки, неспособные синтезировать мРНК. Синтез белка происходит в клетках-предшественниках, еще содержащих ядро. В результате в зрелых эритроцитах имеется набор ферментативных систем, которые могут активно функционировать лишь некоторое время. «Отмирание» клеток сопровождается постепенной потерей активности ферментов и происходит после циркуляции в кровотоке в течение 120 суток. Описан ряд синдромов, обусловленных наследственными повреждениями ферментативных систем эритроцитов. Один из них-несфероцитарная гемолитическая анемия. Генетически обусловленные повреждения ферментов гликолиза. Один из наиболее важных путей катаболизма в зрелых эритроцитах, необходимый для образования богатых энергией фосфатов (АТР),анаэробный гликолиз, или путь Эмбдена—Мейергофа (рис. 4.3). Эта цепь анаэробных реакций приводит к образованию на один моль глюкозы двух молей молочной кислоты и четырех молей АТР, из которых один затрачивается в ходе гликолиза на фосфорилирование глюкозо-6-фосфата и превращение его в фруктозо-1,6-дифосфат и еще один – на превращение глюкозы в глюкозо-6-фосфат. Итого, в полной цепи реакций на моль глюкозы образуется 2 моля АТР, необходимого для разнообразных клеточных функций эритроцитов, таких как поддержание формы (эритроциты представляют собой двояковогнутый диск), работы катионного насоса, а также синтеза разных метаболитов, например глутатиона (GSH) или AMP. Гликолиз катализируется 13 ферментами. Приблизительно 5-10% глюкозо-6-фосфата окисляется на пути так называемого гексозомонофосфатного шунта: в результате последовательности реакций пентозофосфат превращается в фруктозофосфат или глицеральдегид-3-фосфат, которые 16 4. Действие генов
4 Действие генов 17
снова утилизируются в цепи реакций гликолиза. Гексозомонофосфатный шунт – важный источник NADPH, необходимого для восстановления окисленного глутатиона. Эта реакция катализируется глутатионредуктазой. Несфероцитарные гемолитические анемии. В 1953 г. Даше с соавторами описали группу заболеваний, родственных гемолитической анемии, которые назвали несфероцитарными в отличие от наследственного сфероцитоза [1049]. Больные страдали повышенным гемолизом, сопровождавшимся желтухой разной степени тяжести, небольшим увеличением селезенки и образованием камней в желчном пузыре. Эти признаки отличали описанное ими заболевание от наследственного сфероцитоза (18290). Устойчивость эритроцитов к осмотическому давлению у больных несфероцитарной анемией оказалась нормальной, не было обнаружено и структурных изменений гемоглобина. С помощью тонких методов гематологического анализа установлено, что заболевание гетерогенно по своей природе, хотя наблюдается заметное перекрывание параметров для различных форм болезни. Для детального изучения этой группы нарушений необходимо дальнейшее развитие методов энзимологии. Повреждения ферментов гликолиза. В период между 1961-1975 гг. были описаны генетически обусловленные нарушения 11 из 13 ферментов гликолиза. По меньшей мере для 8 из описанных дефектов удалось показать связь с несфероцитарной гемолитической анемией. В ряде случаев наблюдали сопутствующие нарушения центральной нервной системы и мышц. В общем случае уменьшение активности фермента ниже критического значения приводит к накоплению метаболита, предшествующего данному блоку, и к падению концентрации метаболита, образующегося в данной реакции. Недостаточность некоторых из этих ферментов сопровождается побочными эффектами, например снижением уровня АТР. Однако системе присуща способность к регуляции, которая увеличивает ее стабильность, поэтому на основании данных только клинического и гематологического анализа нельзя судить о природе и степени повреждения фермента. Кроме того, обычно подобный анализ проводят на популяции в основном молодых эритроцитов, в которых активность ферментов, как правило, выше, чем в старых клетках, и поэтому недостаточность по отдельным ферментам может остаться незамеченной. Некоторые ферментативные нарушения описаны в таблице 4.3. Нумерация использована та же, что и на рисунке 4.3. Приведенные примеры позволяют сформулировать ряд замечаний более общего характера, касающихся дефектов ферментативных систем человека. Доступность материала для исследования ферментов гликолиза. В настоящее время наследственные повреждения известны почти для всех ферментов гликолиза. Этим гликолиз выделяется среди прочих путей метаболизма, для которых далеко не всегда известно, существуют ли наследуемые дефекты, затрагивающие хотя бы некоторые из ферментов. Проще всего можно объяснить этот факт тем, что необходимую для исследований кровь больных сравнительно легко получить: анализ венозной крови больных, находящихся в стационаре, вполне доступен в отличие, например, от соскоба кожи, не говоря уже о биопсии мозга. Кроме того, эритроциты - это высокоспециализированные клетки, поэтому в них функционируют далеко не все ферментативные системы, имеющиеся в других клетках. Таким образом, количество реакций, которые могут быть нарушены, относительно невелико. Это значительно облегчает анализ. Перечисленные преимущества клеток крови как объекта исследований широко использовались, например, при изучении глюкозо-6-фосфат—дегидрогеназы и особенно в исследованиях гемоглобина. В результате этих работ сложились основополагающие представления о взаимоотношениях генов и белков, которые они кодируют (разд. 4.3), а также о естественном отборе в человеческих популяциях (разд. 6.2.1.6). 18 4. Действие генов
4. Действие генов 19
Энзимологические исследования позволяют выявить генетическую неоднородность. В разд. 3.3 уже говорилось о том, что все попытки выявить генетическую неоднородность популяции на основе изучения фенотипа наталкиваются на непреодолимые препятствия. Если два фенотипа очень сильно перекрываются и характеризуются аутосомно-рецессивным типом наследования, то единственным доказательством их генетической неоднородности может быть рождение у пары гомозиготных родителей только здоровых детей (разд. 3.1.3). Но, если проведен энзимологический анализ, генетическая неоднородность может быть однозначно установлена по следующим признакам. 1. Все наследуемые дефекты ферментов гликолиза в эритроцитах, приведенные в табл. 4.3 и на рис. 4.3, вызывают практически неразличимые по клиническим проявлениям варианты гемолитической анемии. Один из источников генетической неоднородности - сходство или даже совпадение фенотипических проявлений мутаций в генах, кодирующих разные ферменты данного пути метаболизма. Тот же вывод можно сделать, анализируя информацию, полученную при сравнении изученных случаев болезни накопления гликогена. 2. Второй источник неоднородности – разнообразие изменений одного и того же конкретного фермента, вызываемых различными мутациями в его гене. Чем больше методов используют для анализа, тем больше удается обнаружить различий. Вероятность генетической гетерогенности очень велика, поскольку очень велико число мутаций, вызывающих аминокислотные замены. При большинстве наследуемых дефектов ферментных систем у гомозигот сохраняется остаточная активность фермента. Во втором столбце табл. 4.3 представлены значения активности ферментов гликолиза в эритроцитах больных, гомозиготных по наследственным дефектам гликолиза. Во всех случаях, когда удавалось провести измерения, наблюдалась остаточная активность, иногда довольно значительная. В некоторых случаях причиной этого могла быть и активность другого фермента, способного катализировать ту же реакцию. Однако, как правило, повреждения фермента в результате отдельной мутации не столь значительны, чтобы полностью его инактивировать. По мнению Киркмана [1866], такая остаточная активность характерна для большинства наследственных повреждений ферментов у человека. Между тем в клетках бактерий мутации, как правило, вызывают полный блок того или иного пути метаболизма. Это обстоятельство можно объяснить отчасти тем, что в человеческой популяции происходит жесткий отбор на жизнеспособность: полный блок ключевых реакций метаболизма с большой вероятностью оказывается летальным. С другой стороны, у бактерий мутации удается обнаружить главным образом в тех случаях, когда активность того или иного фермента утрачена практически полностью. Мутанты с неполным блоком (leaky) нередко выживают даже на минимальной среде. Существует принципиальная разница между характером проявления мутаций у бактерий и человека. Мутантные штаммы бактерий наиболее часто обнаруживают по утрате способности расти на среде, стандартной для конкретной культуры, в то время как большинство мутаций, затрагивающих ферменты человека, обнаружены у больных наследственными заболеваниями. С появлением методов, позволяющих изучать все типы мутаций у бактерий, у них был идентифицирован полный набор мутаций, сходных со структурными мутациями, затрагивающими ферментные системы человека. Клинические проявления наследственного дефекта фермента тесно связаны с нормальной активностью фермента в различных тканях. В одном организме и даже в отдельной клетке может существовать несколько форм данного фермента [120]. Это так называемые изоферменты. Их существование можно объяснить как негенетическими причинами (вторичные изменения фермента в тканях), так и генетическими (наличием разных генетических локусов, кодирующих полипептиды с более или ме- 20 4 Действие генов
нее значительными структурными различиями). Возможно, для таких пептидов существовал некогда общий предок, и различия между ними – результат дивергенции (разд. 7.2.3). Термин «изоферменты» применяется также к аллельным вариантам одною генетического локуса, различающимся по электрофоретической подвижности. Для многих ферментов показано существование таких множественных форм. Изоферменты катализируют одну и ту же метаболическую реакцию, однако, как правило, они тонко адаптированы к условиям, варьирующим в различных тканях. Наследственные повреждения ферментов - результат мутаций в отдельных генах. Именно поэтому обычно повреждается только один из имеющихся в организме изоферментов данной группы. Если изменения затрагивают более чем один изофермент, то скорее всего имеется общая для них полипептидная цепь или происходят какие-то вторичные изменения структуры фермента. Если поврежденный мутацией фермент активен лишь в одной, но не во многих тканях, фенотипическое проявление такой недостаточности имеет характерные особенности. Действие мутаций может быть плейотропным, т. е. одна мутация может вызывать целый ряд последствий. Естественно предполагать, что недостаточность ферментов, активных в нескольких типах тканей, будет обладать плейотропным эффектом. Это один из вероятных, но, конечно, далеко не единственный механизм возникновения плейотропии. Даже если фермент функционален только в одной ткани, его отсутствие может вызвать изменения в других тканях за счет сдвигов метаболизма, вызванных первичной мутацией. С другой стороны, в некоторых случаях, наследственные дефекты ферментных систем, которые можно обнаружить во всех тканях, выражаются в четких изменениях фенотипа только за счет соответствующих нарушений в одной из них. Вероятно, в других тканях дефект каким-то образом компенсируется. Примеры, перечисленные в табл. 4.3, наглядно иллюстрируют все случаи плейотропного действия мутаций. Например, недостаточность фосфофруктокиназы (PFK) вызывает симптомы умеренной несфероцитарной гемолитической анемии средней тяжести, такие как небольшая анемия, умеренная желтуха, а также некоторое увеличение селезенки: эти изменения фенотипа можно объяснить сокращением времени жизни эритроцитов. Для таких больных характерна нормальная активность PFK в лейкоцитах, тромбоцитах и скелетных мышцах. Проявление этого дефекта, по всей вероятности, ограничено только эритроцитами [1147]. Больные из других семей страдают слабой формой гемолитической анемии, но в сочетании с выраженной миопатией [1191]: как те, так и другие симптомы, являются следствием болезни накопления гликогена [1327]. PFK из мышечных клеток и эритроцитов различаются по электрофоретической подвижности, хроматографическим и иммунологическим характеристикам. Спектр изоферментов, по-видимому, чрезвычайно многообразен: только в эритроцитах обнаружено по меньшей мере два варианта [1148]. Различия в проявлениях плейотропного эффекта в разных семьях могут объясняться тем, что в них наследуются разные мутации, затрагивающие разные полипептидные цепи, которые могут входить или не входить в состав изофермента, адаптированного к конкретной ткани. Однако в ряде случаев ферментативная недостаточность наблюдается во всех исследованных тканях, а изменения фенотипа обнаруживаются только в одной из них. Примером может служить наследственная недостаточность глюкозофосфатизомеразы (GPI). У обследованных больных с пониженной активностью этого фермента в эритроцитах снижение активности имело место также в лейкоцитах, тромбоцитах, фибробластах, мышцах и в печени. Во всех перечисленных тканях биохимические характеристики фермента оказались идентичными. Несмотря на тщательный анализ, обнаружить даже намек на какие-либо тканеспецифические формы не удалось. Тем не менее среди наблюдаемых клинических симптомов 4. Действие генов 21
доминируют те, которые определяются нарушениями функций эритроцитов. Больные страдают тяжелой формой гемолитической анемии, которая проявляется у новорожденных как сильная желтуха. В настоящее время известно довольно много структурных мутаций глюкозофосфатизомеразы: нередко встречаются составные гетерозиготы (компаунды) по таким мутациям. Заметим, однако, что далеко не все случаи недостаточности приводят к возникновению заболевания или сколько-нибудь заметным изменениям фенотипа [1003]. Недостаточность пируваткиназы (РК) (26620). Недостаточность пируваткиназы – один из наиболее часто встречающихся дефектов гликолиза эритроцитов. У больных, гомозиготных по этому нарушению, можно наблюдать самые разнообразные гематологические симптомы. У некоторых из них гемолитическая анемия может быть полностью компенсирована, другие страдают тяжелыми повторяющимися приступами несфероцитарной анемии. Приведем обязательные признаки этого наследственного заболевания: 1) наличие остаточной активности фермента – 5-20% для гомозигот и около 50% для гетерозигот по данному признаку. Клинических признаков заболевания гетерозиготы не обнаруживают; 2) количественные характеристики фермента, такие как кинетика реакции, специфичность связывания с ADP и UDP, термостабильность, оптимальные значения рН и изоэлектрическая точка, указывают на существование целого ряда вариантов с различными свойствами. Вероятно, большинство из них возникло в результате структурных мутаций [1149; 1148]. По результатам анализа на белковом уровне трудно с достоверностью определить, действительно ли больной гомозиготен по данному варианту или является носителем двух дефектных аллелей (компаунд-гетерозигота) (разд. 3.1.3, рис. 3.12). Активность ферментов и клинические проявления у гетерозигот. Для большинства случаев нарушений гликолиза, приведенных в табл. 4.3, уровень активности фермента определяли у больных, гетерозиготных по наследственному дефекту. Как правило, их активность ниже нормальной, но выше, чем у гомозиготных больных. Этот факт отражает общее правило: у гетерозигот по дефектному ферменту сохраняется до 50% нормальной активности. Обычно такое снижение активности не приводит к патологическим симптомам: половины активности вполне достаточно, чтобы поддерживать нормальные функции организма. Тот факт, что гетерозиготы сохраняют не более 50% нормальной активности, очевидным образом подтверждает, что количество фермента жестко контролируется соответствующим локусом. У здоровых гомозигот, несущих два нормальных структурных гена, активность фермента составляет 100%, а у гетерозигот – только 50%. В гетерозиготном организме единственный полноценный структурный ген не в состоянии компенсировать функции мутантного гена, продукт которого неактивен. Это обстоятельство чрезвычайно важно для понимания механизмов генной регуляции у млекопитающих, отличающихся от аналогичных механизмов у бактерий. Аэробное образование энергии в эритроцитах: гексозомонофосфатный путь [994]. В левой части рис. 4.3 представлена цепь аэробных реакций, так называемый гексозомонофосфатный цикл, известный также, как пентозофосфатный цикл, или шунт. Основное его назначение-формирование восстановительного потенциала клетки в виде NADPH. В ходе реакции, катализируемой глюкозо-6-фосфат—дегидрогеназой, происходит окисление глюкозо-6фосфата с образованием 6-фосфоглюконата [1030], который в результате ряда последовательных этапов превращается в О-рибозо-5-фосфат. Ферментативное восстановление окисленного глутатиона сопровождается окислением NADP. Восстановленный глутатион несет SH-группы и может предохранять клетку от действия свободных радикалов таких соединений, как Н2О2. 22 4. Действие генов Обнаружено наследственное заболевание, при котором почти полностью отсутствует восстановленный глутатион. Оно вызвано нарушением синтеза глутатионсинтетазы. В некоторых случаях недостаточность этого фермента сопровождается оксипролинемией [1148]. Недостаток восстановленного глутатиона приводит к развитию несфероцитарной гемолитической анемии, связанной с повышенной чувствительностью к медикаментам - сильным окислителям. Многие лекарства обладают окислительной способностью и снижают восстановительный потенциал клетки [1009; 1269]. Недостаточность глюкозо-6-фосфат — дегидрогеназы (G6PD) (30690) [1079; 999; 1002; 1225; 1224; 1146] (рис. 4.3). Генетически детерминированные нарушения гексозомонофосфатного пути обусловливают повышенную чувствительность к некоторым лекарствам. Это наблюдение лежит в основе одного из ключевых принципов фармакогенетики (см. разд. 4.5.1). Во время войны в Корее 1950-1952 гг. все американские солдаты проходили профилактический курс лечения противомалярийным препаратом «примахин» [8-(4,амино1-метилбутиламино)-6-метоксихинолин]. Примерно у 10% чернокожих солдат и у 1-2 из 1000 белых (в основном выходцев из Средиземноморских стран) в ответ на прием примахина развивалась сосудистая гемолитическая реакция. Ранее сходные реакции иногда наблюдали при лечении сульфаниламидами и памахином больных негров. Было известно также, что гемолитические реакции могут иметь место у некоторых жителей Сардинии после употребления в пищу бобов Vicia faba. Тщательные исследования «примахин-чувствительных» эритроцитов у негров показали, что чем больше возраст эритроцитов, тем выше их чувствительность к гемолизу. Именно это объясняет малую продолжительность гемолитической реакции (рис. 4.4). После гибели всех
4. Действие генов 23
старых клеток популяции гемолиз прекращается, несмотря на продолжение приема примахина. Этот факт вначале пытались объяснить действием иммунных механизмов. Позже была обнаружена нестабильность глутатиона в эритроцитах больных, чувствительных к примахину, при инкубации с ацетилфенилгидразином. В 1956 г. Карсон с сотрудниками обнаружили наследственное повреждение фермента, вызывающее эту нестабильность [1030]. Были изучены следующие реакции (см. также рис. 4.5):
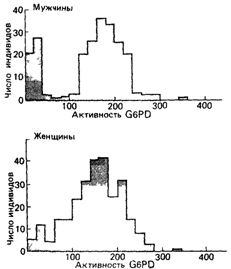
Оказалось, что лимитирующим фактором является недостаточность G6PD, и гемолитические реакции у больных, чувствительных к примахину, связаны с недостаточностью именно этого фермента. Довольно скоро выяснилось, что гемолитические реакции такого типа встречаются чаще у мужчин, чем у женщин. Было проведено количественное определение стабильности глутатиона, основанное на измерении его концентрации до и после инкубации эритроцитов с ацетилфенилгидразином 1). Кривые распределения, построенные для 144 обследованных американских негров, имели ярко выраженный бимодальный характер, причем в значительной части популяции уровень содержания глутатиона был крайне низким. В группе из 184 негритянок кривая смещена влево, а доля больных с низким содержанием глутатиона гораздо меньше, чем в группе мужчин. Отсюда следует, что данный признак сцеплен с Х-хромосомой; низкое содержание глутатиона после инкубации с ацетилфенилгидразином характерно для гомозиготных женщин и гемизиготных мужчин, а промежуточное – для гетерозиготных женщин. Это предположение вскоре получило подтверждение в работах по анализу родословных [1034]. Сходные картины распределения были получены и при использовании методов прямого анализа ферментов в популяции. Заметим, что величины, полученные для гетерозигот, оказались средними между нормой и значением, характерным для гомозиготных больных (рис. 4.6).
1) Этот тест использовали для выявления чувствительности к примахину до обнаружения наследственных дефектов G6PD. 24 4 Действие генов
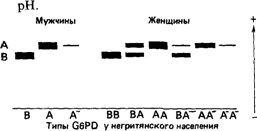
Различия между африканским и средиземноморским вариантами. Спустя несколько лет после открытия недостаточности глюкозо-6-фосфат—дегидрогеназы было обнаружено, что африканский и средиземноморский варианты различаются по степени тяжести детерминируемой ими патологии у мужчин. Активность фермента в эритроцитах негров составляет 10-20% от нормы, тогда как у представителей средиземноморской популяции она никогда не бывает выше 5%. Изучение активности G6PD в лейкоцитах показало, что у негров она практически не отличается от нормы, а у жителей Средиземноморья в некоторых случаях существенно снижена. Для выявления различных форм G6PD использовали метод электрофореза. Подвижность нормального фермента дикого типа обозначили как В. Среди негров с нормальной активностью G6PD у 20% был обнаружен более быстрый компонент: его обозначили как А. У негров с недостаточностью глюкозо-6-фосфат—дегидрогеназы этот фермент обладал подвижностью А-типа, а его активность была сильно снижена (фенотип А–). При недостаточности G6PD, характерной для средиземноморской популяции, электрофоретическая подвижность фермента близка к нормальной (фенотип В–). В популяции здоровых белых глюкозо-6-фосфат—дегидрогеназа практически во всех случаях мигрирует в геле как нормальный компонент В (рис. 4.7). Характеристика различных вариантов G6PD. В популяциях человека выявлен целый ряд редких типов G6PD. В связи с этим встал вопрос о необходимости стандартизации приемов их классификации Рекомендации группы специалистов в этой области были опубликованы Всемирной организацией здравоохранения в 1967 г. [996]. В соответствии с ними характеристика G6PD включает следующие аспекты: а) активность фермента; б) электрофоретическая подвижность в различных буферных системах; в) субстратная специфичность (константа Михаэлиса—Ментен) к глюкозо-6фосфату, NADP и NAD; г) использование аналогов субстрата: 2-дезокси-глюкозо-6-фосфат, галактозо-6-фосфат и деамино-NADP. Аналоги субстрата обычно применяют для выявления тонких качественных различий в свойствах ферментов; д) термостабильность; е) зависимость активности фермента от 4 Действие генов 25
Варианты фермента, характерные для разных популяций человека. На основании перечисленных выше параметров в настоящее время различают более 200 вариантов G6PD [1001] (см. также разд. 7.5.8). Их можно разделить на следующие группы (табл. ААА, ААБ): а) варианты с повышенной активностью фермента. Известно всего дваG6PD Hektoen и G6PD Hartford; б) варианты с активностью, близкой к норме Один из них – упоминавшийся ранее вариант А+ – обнаруживается у 20-25% мужчин-негров в тропической Африке и у их американских потомков; в) варианты с умеренно сниженной активностью. Активность фермента у гемизиготных мужчин составляет от 10 до 50% от нормальной. Иногда обнаруживается чувствительность к лекарствам, вызывающим гемолитические реакции; кожных реакций нет; г) варианты с резко выраженной недостаточностью G6PD и умеренным компенсированием гемолитических реакций. Типичный представитель этой группы – средиземноморский вариант; д) варианты с резко выраженной недостаточностью фермента, сопровождаются хроническим гемолизом даже в 26 4. Действие генов
отсутствие окисляющих агентов. Такие варианты вызывают врожденную несфероцитарную гемолитическую анемию. Приведенная здесь классификация учитывает не все важные особенности глюкозо-6-фосфат—дегидрогеназы, однако она весьма полезна как отправная точка для дальнейших исследований. Углубленный биохимический и молекулярный анализ [1167; 1001; 1002]. Все исследованные случаи недостаточности по G6PD, для которых проводили анализ родословных, подтверждают сцепление гена, детерминирующего этот признак, с Х-хромосомой. Весьма вероятно поэтому, что мутации, обусловливающие все изученные варианты, действительно принадлежат одному локусу и что по крайней мере для эритроцитов не существует другого локуса, кодирующего глюкозо-6-фосфат—дегидрогеназу. Все известные на сегодняшний день варианты, по-видимому, обусловлены различными мутациями в одном структурном гене. Активный фермент имеет молекулярную массу 120 кДа и представляет собой димер. Полипептидные цепи субъединиц состоят из 450 аминокислот: определена их последовательность [1368]. В результате построения пептидных карт после обработки трипсином (метод «отпечатков пальцев») выяснилось, что молекулярные нарушения по меньшей мере двух вариантов заключаются в замене всего одной аминокислоты: G6PD А+ отличается от G6PD В+ единственной заменой аспарагиновой кислоты на аспарагин, а в варианте G6PD Hektoen гистидин замещен на тирозин. Согласно генетическому коду, эти замены вполне могут быть связаны с точковыми заменами оснований в кодирующей цепи ДНК. Таким образом, генетический анализ был перенесен на уровень ДНК (разд. 3.6). Ферментативная активность G6PD обнаружена в клетках большинства (возможно, и всех) тканей; оказалось, что тканеспецифичных форм изоферментов нет, и если имеется мутантная форма глюкозо-6-фосфат—дегидрогеназы, то обнаруживается она во всех тканях. Важность изучения вариантов G6PD для понимания механизмов недостаточности ферментативных систем у человека. Система G6PD служит замечательной моделью, поскольку у мужчин с мутацией в Х-хромосоме имеется продукт только мутантного гена. Напротив, у гетерозигот по аутосомным мутациям нормальный и измененный продукт представлены в соотношении 1:1, и, следовательно, обнаружить незначительные изменения физико-химических свойств продуктов мутантного гена достаточно сложно. G6PD обладает и некоторыми другими особенностями, позволяющими проводить генетический анализ гораздо более подробно, чем это возможно для большинства наследственных дефектов ферментативных систем человека. С помощью этой модельной системы были установлены закономерности, общие для многих наследственных дефектов ферментов человека. 1. Использование широкого набора методов позволяет обнаружить большое количество мутантов, различающихся по ряду параметров. По-видимому, почти каждое вызванное мутацией изменение в структуре фермента влияет на его физиологические особенности. 2. Изменения фенотипа, вызванные мутациями, образуют непрерывный ряд от вариантов с практически неизмененными биологическими функциями (их можно обнаружить лишь с помощью специальных методов) к тем, которые проявляются только в неблагоприятных условиях, и вплоть до вариантов, вызывающих развитие заболевания даже в отсутствие неблагоприятных факторов. Большинство мутаций безразлично для организма и не приводит к развитию болезни. Естественно, что мутации, вызывающие патологические симптомы, обнаруживаются с большей вероятностью, так как больных, страдающих гемолитической анемией, обследуют на предмет недостаточности какого-либо фермента гораздо чаще, чем здоровых людей. 3. Большинство вариантов наследственных нарушений ферментов встречается сравнительно редко. Однако в отдельных популяциях какой-то аллель может 4. Действие генов 27
оказаться распространенным (табл. 4.4); причины этого явления обсуждаются в разд. 6.2.1.6. 4. Практически все мутационные варианты обеспечивают остаточную активность фермента, и, если принять во внимание качественные различия в свойствах ферментов, все типы нарушений можно объяснить структурными мутациями в гене, который удалось точно локализовать в Xхромосоме (разд. 3.4.3). Перечисленные выводы вполне справедливы для большинства или даже для всех наследственных дефектов ферментативных систем человека. Последний вывод связан с локализацией гена G6PD в Х-хромосоме. Известно, что в большинстве клеток гетерозиготных женщин у Х-сцепленных генов функционально активен только один из двух аллелей. Это обстоятельство может оказаться полезным для решения проблем, связанных с ростом опухолей и клеточной дифференцировкой. Так, например, было обнаружено, что в клетках лейомиомы матки у женщин, гетерозиготных по двум электрофоретическим вариантам G6PD, присутствует только один тип фермента [1002]. Это можно объяснить происхождением всех клеток опухоли от одной клетки. Подобные наблюдения, позволяющие предполагать моноклональное происхождение опухолей, имеются для большинства неопластических процессов (см. разд. 5.1.6). Другая проблема связана с количеством стволовых кроветворных клеток, которые дают начало популяции эритроцитов. Если женщина гетерозиготна и имеет, например, аллели G6PD А+ и В +, а инактивация Х-хромосомы происходит случайным образом, то относительное количество стволовых клеток с функционирующими генами А+ и В+ описывается биноминальным распределением: (1/2А+ + 1/2В +) n, где иколичество стволовых клеток. Следовательно, вероятность того, что у гетерозиготы все эритроциты будут А+ (или все В+), составляет (1/2)". На практике для использования этого принципа требуется количественная оценка соотношения А- и В-форм фермента в исследуемой ткани. Предварительные данные свидетельствуют о том, что количество стволовых кроветворных клеток в момент инактивации Х-хромосомы, по-видимому, невелико (на 12-16-й день развития эмбриона всего около 5000 клеток; разд. 2.2.3.3). Затем их количество возрастает, но не более чем в 3-5 раз. Этот метод основывается на том допущении, что отбора клеток с одной из форм G6PD не происходит. Однако имеются некоторые данные, указывающие на наличие селективного преимущества клеток с нормальным вариантом фермента у гетерозигот по гену G6PD. В такой ситуации оценка пула эмбриональных или стволовых клеток становится менее надежной. Фенокопия наследственного дефекта фермента: недостаточность глутатионредуктазы [1077; 1078]. С гексозомонофосфатным циклом тесно связана реакция восстановления GSSG до GSH, которая катализируется глутатионредуктазой (рис. 4.3). В литературе прежних лет о,писан ряд родословных с мнимыми дефектами этого фермента; в таких семьях были обнаружены разнообразные гематологические отклонения. Однако результаты анализа родословных в действительности не согласуются с предположением об обычном дефекте глутатионредуктазы. Позже было показано, что недостаточность этого фермента почти всегда связана с отсутствием его кофермента – флавинадениндинуклеотида, что в свою очередь объясняется низким содержанием в пищевых продуктах рибофлавина (витамина В2) [997]. После введения рибофлавина активность глутатионредуктазы нормализуется в течение нескольких дней. Установлено, что это патологическое состояние часто встречается на севере Таиланда, что обусловлено низким содержанием рибофлавина в традиционной диете. Таким образом, не всякое отклонение в работе фермента является наследственным, даже если оно распространено в определенной популяции. В литературе описаны истинные генетически обусловленные дефекты глутатионредуктазы. У человека известны и другие наследственные отклонения, связанные с ненормально высокой потребностью в 28 4. Действие генов
определенных коферментах, которые необходимо в таких случаях добавлять к пище в виде витаминов. В качестве примера можно привести наследуемый сцепленно с Х-хромосомой рахит, устойчивый к витамину D [958], и зависимость от пиридоксаля, сопровождающаяся эпилептическими припадками (см. также разд. 3.1.4). Возможно, в будущем будет обнаружена недостаточность по глутатионредуктазе, обусловленная наследственной недостаточностью по рибофлавину. Имитация генетического дефекта воздействием извне называется фенокопией. Этот термин был предложен в 1935 г. Гольдшмидтом [1106]. Согласно определению, фенокопия - это воспроизведение генетически детерминированного признака под действием внешних факторов. Вызывая тепловой шок у дрозофилы дикого типа на разных стадиях ее развития, Гольдшмидту удалось получить многочисленные фенотипические отклонения, подобные вариантам, возникающим обычно в результате мутаций. Эксперименты с фенокопиями были выполнены на многих видах, они сыграли важную роль в развитии представлений о механизмах нормального эмбрионального развития и возникновения уродств. Однако их значение было переоценено. Тем не менее при нарушениях метаболизма у человека возможность фенокопии всегда следует проверить, поскольку в таких случаях бывает возможна эффективная терапия. Среди метаболических нарушений человека частой фенокопией является гипотиреоз, обусловленный недостатком неорганического йодида, – патологическое состояние, распространенное в альпийских областях Европы и в некоторых других районах мира. В этом случае недостаточность жизненно необходимого неорганического иона приводит к тем же последствиям, что и нарушение синтеза тиреоидного гормона, которое наблюдается в некоторых семьях (рис. 4.24). Тем не менее, хотя клинически эти случаи близки, с точки зрения патофизиологии, между ними нет ничего общего. Нарушения метаболизма нуклеотидов. В эритроцитах необходимый уровень АТР поддерживается благодаря реакциям гликолиза (рис. 4.3). Известна и другая реакция, ведущая к образованию АТР:
Затем ADP в ходе гликолиза превращается в АТР. Одна из энергоемких функций клетки, для которой необходим АТР,это транспорт катионов. Он непосредственно связан с ферментативной активностью АТРазы. Этот фермент локализован в клеточной мембране и присутствует в двух фракциях, которые могут активироваться ионами К+ и Na+ или Mg++. Он гидролизует АТР до AMP. Недостаточность АТРазы также может вызывать несфероцитарную гемолитическую анемию (НСГА). Описан ряд случаев, несколько различающихся по симптомам и гематологическим характеристикам [1046; 1118; 1120; 1203]. Недостаточность по аденилаткиназе (АК) (20160) зафиксирована у тринадцатилетнего мальчика, страдающего НСГА. Активность АК в эритроцитах составляла 1-13% нормальной. Уровень АТР и ADP в эритроцитах оказался нормальным, а уровень AMP повышен. Дефект наследовался как аутосомно-рецессивный признак. Недостаточность по пиримидин5'-нуклеотидазе была впервые обнаружена у больного, страдающего хроническим гемолизом средней тяжести [1333], а несколько позже у ряда других больных [1148]. Зрелые эритроциты обладают способностью синтезировать NAD в больших количествах из никотиновой кислоты, АТР и 5-фосфо-D-рибозо-1-пирофосфата. Для этой реакции требуется нормальная работа гликолитических ферментов. Гексозомонофосфатный цикл, по-видимому, не является существенным источником рибозофосфата, поскольку в клетках, дефектных по G6PD, содержание NAD соответствует норме. Для синтеза NADP из NAD нужна киназа. Возможно, сравнительно небольшое число установленных нарушений метаболизма нуклеотидов не отражает действительной ситуа- 4 Действие генов 29
ции, оно может объясняться сложностью методов определения активности ферментов. Изучение ферментов, не функционирующих в клетках крови, а также соответствующих дефектов связано с серьезными трудностями. Многие такие ферменты удается обнаружить в фибробластах, выращенных в культуре после биопсии кожи. В отличие от эритроцитов фибробласты содержат ядра. Они способны делиться и осуществлять все стадии синтеза белков и потому значительно полнее, чем эритроциты, обеспечены ферментами. Фибробласты лишены лишь некоторых ферменгов, характерных для специализированных групп клеток, например клеток печени (в частности, в фибробластах нет фенилаланингидроксилазы, которая является дефектной при фенилкетонурии). В фибробластах выявляются нарушения ферментов, катализирующих многие различные метаболические пути. Именно поэтому изучение активности ферментов в фибробластах внесло существенный вклад в наши знания о дефектах ферментов. Ниже мы остановимся на одной группе заболеваний, которая также позволяет сформулировать ряд более общих выводов о дефектах ферментов у человека. Это мукополисахаридозы, которые относят к обширной группе патологических состояний, обусловленных нарушением различных ферментов лизосом, а также - сфинголипидозы и муколипидозы. Дата добавления: 2015-12-16 | Просмотры: 1590 | Нарушение авторских прав |