|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
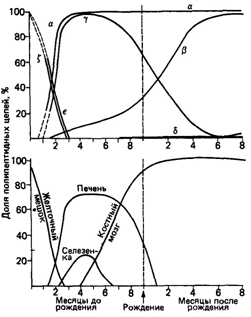
Генетика гемоглобинаМолекулы гемоглобина. Молекула человеческого гемоглобина состоит из четырех полипептидных цепей. Молекула гемоглобина обозначается общей формулой α2β2, которая показывает, что в состав молекулы входят две пары сходных цепей глобина [1348]. Большинство разновидностей гемоглобина человека имеют идентичные α-цепи и различаются по другим цепям. К каждой цепи глобина в специфическом участке присоединяется молекула небелковой природы - гемогруппа, или гем (рис. 4.34). Четыре глобиновые цепи, каждая со своим гемом, образуют функциональную молекулу гемоглобина, которая переносит кислород из легких в ткани. Молекула глобина построена из 140 с небольшим аминокислот, которые расположены в строго определенном порядке (рис. 4.35). Последовательность аминокислот в белке (например, в гемоглобине) считают его первичной структурой. Пространственное расположение соседних остатков называется вторичной структурой, а трехмерное расположение белковых субъединиц - третичной структурой (рис. 4.34). Термин четвертичная структура относится к взаимной организации четырех субъединиц в составе функционирующей молекулы. Преобладающим типом гемоглобина у детей и взрослых является НbА, или гемоглобин взрослых (α2β2). Его отличительная черта - строение Р-цепи (рис. 4.35). α- и β-цепи различаются по многим аминокислотным остаткам. У всех взрослых есть небольшое количество (2-3%) гемоглобина НbА2(α2δ2). Характерная для него δ-цепь отличается от β-цепи только по десяти аминокислотным остаткам. После рождения у всех детей обнаруживается также небольшое количество (меньше 1%) фетального гемоглобина HbF:α2γ2 (см. ниже), γ-цепь значительно отличается от α- и β-цепей. α-цепи НbА, НbА2 и HbF идентичны. Существует несколько типов гемоглобинов, характерных для эмбрионального и фетального развития, ζ-цепи напоминают по аминокислотному составу α-цепи [1155], а ε-цепи похожи на β-цепи [1232]. ζ-цепи, вероятно, появляются раньше других в эмбриональном развитии. ζ- и ε-цепи исче- 4. Действие генов 73
зают через 8-10 недель внутриутробного развития (рис. 4.36) [1364]. Затем преобладающим становится гемоглобин HbF(α2γ2), который отличается от других присутствием γ-цепи. Известно два типа γ-цепей: с аланином (Аγ) или с глицином (Gγ) в 136-м положении. Существует и третий тип γ-цепи с треонином вместо изолейцина в 75-м положении [1281; 1319]. Он встречается у 10-15% эмбрионов и, судя по всему, не связан с каким-либо нарушением. Гемоглобин α2β2 обнаруживается уже на 6-8 неделе развития плода [1319; 1364]. Синтез γ-цепей у эмбриона происходит в основном в печени и селезенке, но могут они синтезироваться и кроветворными клетками костного мозга. Наоборот, β-цепи, в детстве и в более зрелом возрасте синтезируются главным образом в костном мозге, однако синтез вне костного мозга также возможен [1364]. Различные типы гемоглобина перечислены в табл. 4.12. Все нормальные гемоглобины человека, которые были исследованы, имеют идентичную трехмерную структуру (рис. 4.34), существенную для переноса кислорода. Все 74 4 Действие генов
4 Действие генов 75
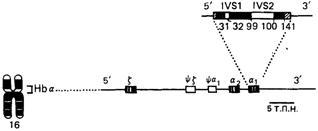
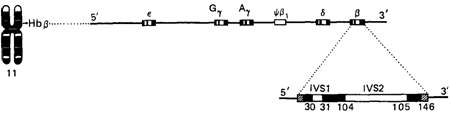
глобиновые цепи различных гемоглобинов имеют общее эволюционное происхождение и возникли в результате последовательных дупликаций генов (см. разд. 7.2.3). Чем больше сходство между двумя цепями, тем позднее в эволюции произошла дупликация. Очевидно, цепи Аγ и Gγ, которые различаются по одной аминокислоте, дивергировали позже всех других, а дупликация генов β- и α-цепей произошла в весьма отдаленном прошлом. Гены гемоглобина. Аминокислотная последовательность каждой глобиновой цепи кодируется своим собственным геном. В гаплоидном наборе у нормального человека присутствует по крайней мере по одному гену α, β, γ, δ, ε, ζ, и по крайней мере по два таких гена-в диплоидном наборе. В большинстве популяций человека ген α-цепи существует в дуплицированном состоянии, причем отличий между двумя α-генами не обнаружено [1350]. Имеются два гена γ-цепей Аγ и Gγ, которые различаются по кодону, детерминирующему аминокислотный остаток в 136-м положении. Некоторые гены Аγ несут необычный кодон, в результате в 75-м положении изолейцин замещен на треонин (TАγ). Синтез небелковой гемогруппы также контролируется генами, поскольку они кодируют ферменты, обеспечивающие биосинтез гема. Различные гены глобинов, соответствующие им глобиновые цепи и различные нормальные гемоглобины приведены в табл. 4.12 и на рис. 4.37. Была подробно исследована структура всех генов глобинов, опубликованы их полные нуклеотидные последовательности [981; 1041; 1200; 1273; 1304; 1314]. Подобно многим генам млекопитающих, гены глобинов у человека образуют мультигенное семейство и расположены на хромосомах в составе двух кластеров (рис. 4.38, 4.39). α-кластер глобиновых генов занимает 25000 пар оснований (25 т. п. н.) в коротком плече 16-й хромосомы. Семейство γ-β-δ-генов глобина расположено в коротком плече 11-й хромосомы на участке длиной 60 т. п. н. Пока остается неизвестным генетический механизм, регулирующий координированное функционирование генов на двух различных хромосомах, в результате которого образуется равное количество субъединиц α- и β-у-типа. В α-кластере структурные гены расположены в следующем порядке в направлении от 5' к 3': ген эмбриональной ζ-цепи, псевдоген ζ-цепи,
76 4. Действие генов
псевдоген α-цепи и два идентичных гена α-цепи (рис. 4.38). Выяснено расположение генов и в β-кластере: ген эмбриональной ε-цепи, два гена фетальных γ-цепей (Аγ и Gγ), псевдоген β-цепи, ген δ-цепи и ген β-цепи (рис. 4.39). Порядок расположения генов в этих кластерах совпадает с очередностью их экспрессии в онтогенезе. По последовательности нуклеотидов псевдогены мало отличаются от своих функциональных гомологов. Однако в результате различных мутаций стала невозможной их транскрипция и, следовательно, экспрессия. Предполагается, что псевдогены возникли в результате дупликации, после которой их экспрессия перестала быть необходимой для нормального функционирования организма. Ген δ-глобина, продукт которого составляет лишь 2-3% всего гемоглобина, можно считать геном, который находится в переходном состоянии к псевдогену. Все глобиновые гены во многом сходны по своей функциональной организации. Каждый из них имеет в составе три кодирующие последовательности, т. е. три экзона. Между 1-м и 2-м экзонами и между 2-м и 3-м экзонами расположены уникальные вставочные последовательности, или интроны, известные соответственно как IVS-1 и IVS-2 (от англ. intervening sequences) (рис. 4.38, 4.39, 4.40). Интроны транскрибируются вместе с экзонами, так что в первичном транскрипте представлены как кодирующие, так и некодирующие последовательности соответствующего гена. Вставочные последовательности вырезаются в ходе процессинга, который происходит в ядре, в результате конец первого экзона соединяется с экзоном 2, а конец второго экзона - с экзоном 3, при этом образуется функциональная мРНК, которая и служит матрицей для синтеза гемоглобина на рибосомах (рис. 4.40). Две вставочные последовательности идентичны у различных генов γ-δβ-кластера, но отличаются от более коротких интронов генов α-кластера. Детали процесса сплайсинга пока не ясны, однако для его изучения оказались весьма полезными мутации, которые вызывают β-талассемии (см. ниже) и обусловлены нарушениями вырезания интронов. Все интроны начинаются с нуклеотидов GT (донорный сайт) и кончаются динуклеотидом AG (акцепторный сайт) - эти динуклеотиды состав- 4. Действие генов 77
ляют часть так называемых обобщенных последовательностей сайтов сплайсинга. Более подробно см. в [1041 и 1238]. Некоторые детали этапов синтеза гемоглобина (от гена до белковой молекулы) представлены на рис. 4.40 и 4.41. Генетические доказательства несцепленности генов α- и β-глобинов появились задолго до определения структуры кластеров этих генов. Было показано, что если один из родителей является двойной гетерозиготой с мутациями в генах α- и β-глобинов, а другой - нормальной в отношении гемоглобина гомозиготой, то в потомстве выявляются четыре фенотипа: нормальный, с измененным α-глобином, с измененным β-глобином и двойной мутант (рис. 4.42)
78 4. Действие генов
[1014]. Если бы гены α- и β-глобинов были тесно сцеплены, то в потомстве наблюдались бы генотипы Hbαx и НbβХ, но не было бы двойных мутантов или нормальных индивидов. Подобным образом генетически доказано тесное сцепление генов δ- и β-глобинов: если один из родителей был двойной гетерозиготой с мутациями в генах β- и δ-цепей, то рекомбинантов среди детей не было [1013] (рис. 4.43). Открытие гемоглобина Lepore - продукта слияния генов δ- и β-цепей - послужило биохимическим доказательством сцепления этих генов в составе одной хромосомы [1350] (см. ниже). Вывод о сцеплении генов γ- и β-глобинов был сделан на основании исследований гемоглобина Kenya, ген которого образуется при слиянии этих двух генов. Промоторы. Перед каждым глобиновым геном расположены три различные последовательности. Они близки по структуре у разных генов и, судя по всему, участвуют в регуляции транскрипции (рис. 4.40). Их называют промоторными элементами [1041; 1238]. В их число входит ТАТА или АТА-блок (последовательность Хогнесса), который находится на расстоянии в 30 пар нуклеотидов от точки начала транскрипции. Эта последовательность представляет собой элемент промотора, необходимый для точной инициации транскрипции. Другая последовательность, СААТ, расположенная за 80 пар оснований от стартовой точки, служит сайтом узнавания для РНКполимеразы. Третий, дистальный, элемент локализован за 80-100 нуклеотидов, имеет характерную последовательность PuCPuCCC (Pu-пурин). До сих пор неизвестно, требуются ли для образования глобинов «энхансеры» (усилители)-генетические элементы, влияющие на эффективность транскрипции независимо от их позиции или ориентации. Последовательности, расположенные за геном. Терминация транскрипции осуществляется примерно через 1000 пар оснований после 3-го экзона гена β-глобина (рис. 4.40). Сигналом расщепления РНК эндонуклеазой служит последовательность AAUAA, к которой затем присоединяется polyA-«хвост» длиной в 220 нуклеотидов. Она не 4. Действие генов 79
закодирована в ДНК и необходима для стабилизации мРНК, которая переносит генетическую информацию от ядерных генов к рибосомам, где в результате соединения аминокислот в нужной последовательности происходит синтез глобинов (рис. 4.41). Полиморфизм ДНК в области глобиновых генов. [972; 1253]. При картировании генов γ-δ-β-кластера с помощью рестрикционного анализа была обнаружена значительная вариабельность последовательности ДНК у различных индивидов (рис. 4.40). Все известные варианты β-глобинового комплекса генов возникли в результате одиночных нуклеотидных замен и обозначаются как присутствующие (+) или отсутствующие (—). Среди 17 полиморфных сайтов в β-кластере 12 локализованы во фланкирующих последовательностях, 3 внутри интронов, 1 внутри псевдогена и только 1 внутри кодирующей части гена р-глобина (синонимическая замена). Такое расположение закономерно, поскольку мутации в кодирующих областях скорее могут вызвать нежелательные эффекты. Большая часть ДНК, расположенной между структурными генами, не экспрессируется, поэтому изменения нуклеотидной последовательности в этих районах обычно не имеют функциональных последствий. Различные полиморфные сайты имеют древнее происхождение, поскольку они обнаружены у всех расовых групп (табл. 4.13). Заметим, однако, что некоторые варианты встречаются только у негров, у других расовых групп их нет. Два случая полиморфизма ДНК в α-глобиновом локусе относятся к гипервариабельным районам, состоящим из различного числа случайно повторенных фрагментов ДНК длиной 36 нуклеотидов (разд. 2.3.3.9). Специфическое сочетание полиморфных сайтов в генном кластере (или генетическом локусе) называется гаплотипом. Например, расположение пяти сайтов возможного полиморфизма можно записать как + – + – + в направлении от 5' к 3'. Совокупность четырех основных гаплотипов, различающихся между собой минорными вариациями в 5 сайтах гена β-глобина, (табл. 4.14) была названа «остов». Отличительной чертой вариабельности ДНК в β-глобиновом кластере является неравновесие по сцеплению полиморфных сайтов. Если бы в течение многих поколений происходила свободная рекомбинация, сочетание полиморфных сайтов было бы случайным, а число различных гаплотипов составило 2n, где n - количество возможных сайтов полиморфизма. В действительности обнаруживается лишь несколько гаплоти- 80 4. Действие генов
пов. Например, имеет место сильное неравновесие по сцеплению восьми сайтов полиморфизма в 5'-фланкирующей области гена δ-глобина (сайты 1-8 на рис. 4.44), вследствие чего 94% всех хромосом в популяции содержит лишь четыре гаплотипа из всех возможных. Сходным образом, для пяти других полиморфных сайтов, локализованных в гене β-глобина и его 3'-фланкирующей области (сайты 12-17 на рис. 4.44), только четыре гаплотипа на участке длиной 18 т.п.н. характеризуют 90% всех хромосом. При сравнении этих двух кластеров полиморфных сайтов неожиданно оказалось, что их сочетания полностью подчиняются случайному распределению. Проще всего это можно объяснить, предположив, что между кластерами имеется горячая точка рекомбинации - участок, в котором рекомбинация происходит с высокой частотой. Такая рекомбинация уже продемонстрирована в одной из семей. Точные границы этой области с высокой частотой рекомбинации пока не определены. Варианты гемоглобинов. Варианты гемоглобина возникают вследствие различных мутационных событий в конкретном глобиновом гене. Чаще всего разные варианты гемоглобина отличаются друг от друга одной аминокислотой в глобиновой цепи. Описано около 350 таких единичных замен [119]. Эти аминокислотные замены вызываются замещением всего одного нуклеотида в триплете. Например, при замене GUA и GAA смысл кодона меняется и место валина в глобиновой цепи занимает глутаминовая кислота (рис. 4.45). Если новая аминокислота отличается от исходной по заряду, измененный гемоглобин будет аномальным по электрофоретическим свойствам. Мутации, которые не влияют на заряд полипептида, обычно удается обнаружить
4 Действие генов 81
только в том случае, если они существенно нарушают функционирование гемоглобина и приводят к болезни. Большинство мутаций гемоглобина независимо от того, меняют они заряд молекулы или нет, не влияют на функции гемоглобина и не приводят к патологии. Как правило, аминокислотные замены в участках полипептидной цепи, которые в молекуле гемоглобина обращены наружу, оказывают меньшее воздей-
ствие на функцию, чем замены аминокислот во внутренних частях цепей или в участках присоединения тема. Замены, нарушающие нормальную спиральную структуру цепи, часто вызывают нестабильность гемоглобина. Замены аминокислот в участках, которыми субъединицы контактируют друг с другом, влияют на сродство к кислороду [1320]. Большинство гемоглобиновых вариантов-редки. Лишь немногие, например гемоглобины HbS, HbC иНbЕ, встречаются чаще других (разд. 6.2.1.6). В кодирующей области гена полиморфизм тоже регистрируется. Известно, что генетический код - вырожденный (табл. 2.12), т. е. несколько триплетов кодируют одну и ту же аминокислоту (см. рис. 4.45). Анализ двух различных замен в 67-м положении цепи β-глобина (рис. 4.45) показал, что два индивида, у которых произошли мутации, и появились новые формы гемоглобина, должны были различаться по исходным триплетам, кодирующим валин в 67-м положении (рис. 4.45). Таким образом, у разных индивидов различные кодоны могут кодировать одну и ту же аминокислоту. Клиническое значение вариантов гемоглобина. Нарушение функций гемоглобина ведет к возникновению различных заболеваний. Существуют четыре основных типа болезней гемоглобина: 1) гемолитические анемии, вызванные нестабильностью гемоглобинов; 2) метгемоглобинемии, обусловлен- 82 4. Действие генов ные ускоренным окислением гемоглобина; 3) эритроцитоз, вызванный нарушением сродства гемоглобина к кислороду и 4) серповидноклеточные нарушения как следствие повреждений клеточных мембран гемоглобином S. Во всех случаях, кроме серповидноклеточных нарушений, гетерозиготы по аномальным гемоглобинам страдают различными заболеваниями, т. е. мутации ведут себя как аутосомно-доминантные. Нестабильные гемоглобины [31; 1335-1357]. Описано свыше 100 нестабильных гемоглобинов. В большинстве случаев мутация затрагивает β-цепь. У многих нестабильных гемоглобинов в полипептидной цепи обнаруживаются аминокислотные замены или делеции в участках связывания гема. Клинические проявления варьируют от едва заметной нестабильности, практически не имеющей клинических последствий, до выраженной нестабильности, при которой происходит интенсивное разрушение эритроцитов. В некоторых случаях гемолиз усиливается при лечении сопутствующих заболеваний сульфониламидами. Нестабильность этих гемоглобинов часто обусловлена преждевременной диссоциацией тема и глобиновой цепи. Такие лишенные гема молекулы глобина преципитуруют внутри клетки, образуя так называемые тельца Хейнца, нарушающие функционирование клеточных мембран. В селезенке тельца Хейнца могут быть удалены из эритроцитов без их разрушения. В конечном итоге такие эритроциты преждевременно уничтожаются ретикуло-эндотелиальной системой. При некоторых формах нестабильности гемоглобина сильный гемолиз удается смягчить удалением селезенки. Точный диагноз нестабильности гемоглобина может быть затруднен, особенно если не наблюдается изменений электрофоретической подвижности. В этом случае необходимо выделение преципитированных глобиновых цепей для дальнейшего анализа в специализированных лабораториях. Нестабильные гемоглобины являются причиной врожденных несфероцитарных гемолитических анемий. Такие гемоглобины могут возникать в результате новых мутаций. Метгемоглобинемия, обусловленная гемоглобином М [31]. Гемоглобин М интересен с исторической точки зрения, так как это первая доминантная гемоглобинопатия, выявленная в 1948 году в семье с врожденным цианозом [1130]. Любопытно, что рецессивная недостаточность метгемоглобин-редуктазы, которая также приводит к метгемоглобинемии, была первым изученным дефектом фермента у человека [1100]. Таким образом, метгемоглобинемия может быть вызвана как доминантной мутацией самого глобина, так и рецессивно наследуемой недостаточностью соответствующего фермента. Известно пять различных мутаций, приводящих к образованию гемоглобина М. Собственно метгемоглобинемия обусловлена ускоренным окислением двухвалентного железа до трехвалентного (табл. 4.15). В четырех случаях образование гемоглобина М вызвано заменой одного из гистидинов, удерживающих группу гема в ее специфическом «кармане» (рис. 4.34) в глобиновой молекуле и стабилизирующих железо гема в его окисленной форме, на тирозин. Пятая мутация - гемоглобин Milwaukee 1 - пока не может быть достаточно четко объяснена с молекулярной точки зрения. Больные с мутациями в α-цепи, вызывающими образование гемоглобина М, страдают цианозом от рождения. При мутации в β-цепи цианоз развивается только через 6
4 Действие генов 83
месяцев после рождения, когда происходит замена γ-цепи на β-цепь. У больных с гемоглобином М обычно наблюдается слабый гемолиз. Эритроцитоз, вызванный образованием гемоглобинов с нарушенным сродством к кислороду [31, 992]. Существует около 30 гемоглобинов с повышенным сродством к кислороду. В 11 случаях мутации происходят в месте контакта α1β1-субъединиц в тетрамере. При адсорбции кислорода происходит движение глобиновых субъединиц в месте контакта между цепями. Повышенное сродство к кислороду может быть вызвано стабилизацией «окси»-конформации или дестабилизацией «дезокси»-конформации (рис. 4.46). Большинство других гемоглобинов с высоким сродством к О2 содержат мутации на СООН-конце β-цепи или в сайтах связывания дифосфоглицерата. В норме эти сайты обеспечивают стабильность «дезокси»-конформации. Повышенное сродство к кислороду приводит к уменьшению количества кислорода, освобождающегося из комплекса с гемом в тканях организма, и вызывает гипоксию (рис. 4.46). Гипоксия ведет к выделению гормона эритропоэтина, стимулирующего образование эритроцитов и собственно эритроцитоз. Больные с эритроцитозом, обусловленным аномалиями гемоглобина, иногда ошибочно диагностируются как больные истинной полицитемией. Однако в отличие от полицитемии при дефектах гемоглобина наблюдается доминантное наследование, а спленомегалия, лейкоцитоз и тромбоцитоз отсутствуют. Известны спорадические случаи подобных дефектов гемоглобина, показано, что они возникли в результате новых мутаций. Было обнаружено всего три гемоглобина с уменьшенным сродством к кислороду [992]. При таком дефекте количество кислорода, поступающее в ткани, увеличивается, поэтому следует ожидать уменьшения образования эритропоэтина. В двух случаях, как и следовало ожидать, наблюдалась слабовыраженная анемия. Серповидноклеточные нарушения [31; 1211; 1298]. Образование гемоглобина S вызвано заменой глутаминовой кислоты на валин в 6-м положении β-цепи. В отличие от всех других замен, эта сильно влияет на растворимость и кристаллизацию гемоглобина в условиях гипоксии. Больные серповидноклеточной анемией наследуют мутантный ген от обоих родителей и не имеют гемоглобина А. При сравнительно низком уровне гипоксии гемоглобин S у таких больных полимеризуется с образованием пучков или волокон. Аномальные кристаллы гемоглобина нарушают структуру мембраны эритроцитов и обусловливают их серповидную форму (рис. 4.47). Некоторые из этих клеток остаются необратимо серповидными и преждевременно разрушаются. Серповидные клетки увеличивают вязкость крови и мешают ее нормальной циркуляции в небольших кровеносных сосудах. Вызванная этим гипоксия приводит к образованию еще большего числа серповидных клеток. Возникает порочный круг, для которого характерны стазы (замедление кровотока) и эпизодические кризы с болями в животе и скелетных мышцах. Через несколько лет пониженное кровоснабжение часто приводит к некрозу органов, например селезенки, что в свою очередь ведет к их атрофии. У гетерозиготных носителей, которые имеют один нормаль- 84 4. Действие генов
ный ген β-глобина НbβА и один мутантный (HbβS), гемоглобин S составляет только 25-40% всего гемоглобина. Клинически такие люди вполне нормальны. Их эритроциты содержат как гемоглобин А, так и гемоглобин S, и по продолжительности жизни не отличаются от нормальных эритроцитов. Серповидноклеточность у таких индивидов сказывается только в условиях сильной гипоксии, например, при нахождении на высоте свыше 3000 м над уровнем моря [1292]. Серповидноклеточность может проявляться слабее, если в организме помимо гемоглобина S имеется другая редкая форма гемоглобина. Присутствие гемоглобина F в эритроцитах больных с серповидноклеточной анемией снижает степень агрегации и кристаллизации гемоглобина S, в результате пациенты, у которых гемоглобин F находится в высокой концентрации, имеют слабовыраженные симптомы серповидноклеточной анемии или не имеют их вовсе. В некоторых случаях присутствие гемоглобина F обусловлено геном, вызывающим постоянный синтез фетального гемоглобина в течение всей жизни (см. ниже). В целом, существует обратная корреляция между количеством гемоглобина F и остротой симптомов серповидноклеточной анемии. Таким образом, любое увеличение количества фетального гемоглобина приводит к ослаблению клинических симптомов серповидноклеточной анемии [970]. Клиническое проявление талассемий будет обсуждаться ниже. 4.3.3. Другие типы мутаций, изменяющих гемоглобин [1188; 1349] Делецш. Установлено, что гены, детерминирующие синтез глобиновых цепей, могут делетироваться. Делеции генов Нbα приводят к а-талассемии, делеция генов Нbδ и Нbβ вызывает наследственное персистирование фетального гемоглобина или Нbδβ-талассемию (см. ниже). Делеция, затрагивающая один триплет нуклеотидов, или один кодон, приводит к выпадению в цепи соответствующей аминокислоты. Делеция четырех кодонов (т.е. 12 нуклеотидов) обусловливает выпадение четырех аминокислот. Были обнаружены делеции протяженностью до 15 нуклеотидов, приводящие к утрате 5 аминокислот (табл. 4.16). По всей вероятности, более крупные делеции приводили бы к потере функциональной активности молекулы гемоглобина. Большинство делеционных гемоглобинов либо нестабильны, либо приводят к увеличению сродства к О2, а во многих случаях имеют оба этих свойства (табл. 4.16). Если число нуклеотидов, утраченных при делеции, не кратно трем, то на участке гена, расположенном за делецией, смысл считываемой генетической информации полностью меняется - в результате возникает совершенно новая аминокислотная последовательность (мутации сдвига рамки считывания). В некоторых случаях образующиеся при этом глобиновые полипептиды удается идентифицировать. Оказалось, что мутация «гемоглобин Wayne» (рис. 4.48) обусловлена делецией одного нуклеотида в 139-м кодоне вблизи конца гена α-глобина, состоящего из 141 триплета. Нуклеотиды терминирующего 142-го кодо- 4. Действие генов 85
на считываются в другой фазе, и новая рамка считывания продолжается до первого в этой рамке терминирующего кодона (UAG). Таким образом, формируется слегка удлиненная цепь молекулы гемоглобина, содержащая 5 дополнительных аминокислотных остатков, которые кодируются 3'фланкирующей областью гена (рис. 4.38 и 4.48). Поскольку рамка считывания сдвинута, эта аминокислотная последовательность отличается от дополнительной аминокислотной последовательности, возника-
86 4 Действие генов
ющей при мутации в стоп-кодоне гена Нbα, например при мутации «гемоглобин Constant Spring» (рис 4 48) В этом случае трансляция происходит без сдвига рамки считывания Совершенно естественно, что делеция, которая приводит к фенотипу «гемоглобин Wayne», локализуется вблизи конца α-цепи Действительно, любая делеция, вызывающая сдвиг рамки считывания на протяженном участке структурного гена, по всей вероятности, будет приводить к синтезу функционально неактивных полипептидов Фенотипически это проявится как «талассемия», и продукт гена вообще не будет обнаруживаться (как при β°-талассемии) По-видимому, возникновение делеций является следствием ошибочного спаривания между гомологичными последовательностями во время мейотического или митотического деления развивающихся генеративных клеток При рассмотрении нуклеотидных последовательностей, окружающих области делеций у различных делеционных мутантов, обнаруживаются участки гомологии, которые могут быть причиной неправильного спаривания Если оно произошло, последующие рекомбинационные события приведут к возникновению делеций различной протяженности Результатом неправильного спаривания может быть и образование комбинированных (или составных) генов Белковые продукты таких генов состоят из N-концевой части одного глобина и С-концевой части другого В качестве примера можно привести гемоглобин Lepore Его синтез контролируется комбинированным геном Нbδ-β
4 Действие генов 87
(рис. 4.49). Известно несколько таких генов, возникающих при кроссинговере в разных точках. Они различаются по относительной длине последовательностей δ- и β-генов, входящих в их состав (рис. 4.49). Гемоглобин Kenya возникает в результате ошибочного спаривания генов НВАγ и Нbβ и последующего кроссинговера. Его хромосома содержит только ген HbGγ и комбинированный ген НbАγ-β (рис. 4.51). Дупликации. Дупликации могут охватывать целые гены. Именно так произошло в ходе эволюции различных цепей глобина. На более поздних этапах при внутрихромосомных дупликациях появились два гена а-глобина и два гена у-глобина (Аγ и Gγ). Известны внутригенные дупликации. Например, при мутации «гемоглобин Grady» в а-цепи глобина дуплицированы остатки 116-118 [1136]. Дупликации одного или двух нуклеотидов могут приводить к мутациям со сдвигом рамки считывания. Подобные мутации обнаружены вблизи конца гена β-цепи [31]. Возникновение гемоглобина Так является следствием дупликации нуклеотидов AG после 146-го кодона, а гемоглобина Cranston - дупликации AG сразу после 144-го кодона в β-цепи (рис. 4.50). В положениях 145 и 146 этого гемоглобина находятся аминокислоты, которые не встречаются в соответствующем участке у других вариантов β-глобина. Гемоглобин Tak имеет нормальную аминокислотную последовательность до 146-й аминокислоты включительно. Нормальная β-цепь содержит 146 аминокислот. Сдвиг рамки считывания при дупликации двух нуклеотидов в случае гемоглобинов Tak и Cranston приводит к возникновению идентичных рамок вслед за 146-м кодоном. Оба мутантных гемоглобина имеют на С-конце добавочные аминокислотные последовательности, которые кодируются нуклеотидами, расположенными непосредственно за нормальным стоп-кодоном. Терминация трансляции в этом случае происходит с участием нового нонсенс-кодона (UAA) в положении 158 (рис. 4.50). Если дупликация одного или двух нуклеотидов происходит внутри гена, а не у 88 4. Действие генов
его конца, рамка считывания нарушается на большом протяжении. Маловероятно, что при этом будет синтезироваться функциональная молекула глобина. Дупликации, как и комбинированные гены, должны возникать в результате неравного кроссинговера (рис. 4.49 и 4.51). Действительно, вариант гемоглобина анти-Lepore встречался несколько раз и был описан под названием гемоглобин Hb Miyada, P-Congo или P-Nilotic (рис. 4.49). Предполагаемый вариант анти-Kenya (gγ, А γ, δ, β-Аγ, δ, β) (рис. 4.51) не был до сих пор обнаружен. Дупликации, так же как и делеции, возникают, по-видимому, вследствие ошибочного спаривания и последующего негомологичного кроссинговера (рис. 4.51).
4.3.4. Талассемии [31; 972; 138; 1253; 222; 97а] Генетически детерминированное снижение или полное подавление синтеза той или иной цепи гемоглобина обусловливает ряд разнообразных патологических состояний, известных под общим названием «талассемии». Это слово происходит от греческого «Таласса» - Средиземное море, первоначально оно отражало средиземноморское происхождение большинства индивидов - носителей генов, ответственных за талассемию. Хотя с точки зрения географии или этнографии этот термин нельзя считать правильным, он используется достаточно широко. Все случаи талассемии можно под-
90 4. Действие генов
разделить на α- и β-талассемии. Талассемии различаются по этиологии, поскольку показано, что ослабление синтеза цепей гемоглобина может быть обусловлено несколькими генетическими механизмами [1037]. Успехи в исследовании талассемий на молекулярном уровне привели к тому, что мутационные повреждения, характерные для этой группы заболеваний, изучены в настоящее время гораздо полнее любых других мутаций у млекопитающих. Изучение различных генов, ответственных за талассемию, позволило многое узнать о структуре, функции и организации глобиновых генов в норме. Выяснилось, что мутации, затрагивающие различные этапы синтеза гемоглобина, могут ослаблять синтез гемоглобина (β+-талассемий) или даже полностью его предотвращать (β0-талассемии) [972; 1253; 1238; 4341] (табл. 4.17, 4.18). Транскрипционные или промоторные мутации. Мутации, которые вызывают талассемию и затрагивают 5'-некодирующую область гена Нbβ, можно рассматривать как регуляторые мутации, влияющие на транскрипцию. Такие мутации обнаружены в константном регуляторном элементе с последовательностью PuCPuCCC и внутри ТАТА-последовательности (табл. 4.17). Эти мутации ослабляют синтез гемоглобина и проявляются как относительно легкие формы талассемий. Мутации, затрагивающие СААТ-сайт, в настоящее время не обнаружены. Мутации в сайте полиаденилирования РНК. У негров часто обнаруживается одиночная мутационная замена ААТААА ® ААСААА в 3-фланкирующей области гена Нbβ, приводящая к β+-талассемий; таким образом, мутации в 3-некодирующей области также могут влиять на эффективность транскрипции. Относительно слабое проявление β-талассемий в этой расовой группе объясняется явным преобладанием мутаций, затрагивающих ТАТА-последовательность (см. выше) и сайт полиаденилирования (табл. 4.17). Нонсенс-мутации и мутации со сдвигом рамки считывания. Как уже указывалось ранее, мутации, приводящие к возникновению терминирующего кодона внутри экзона гена гемоглобина, обусловливают синтез укороченной, неактивной цепи и вызывают поэтому β0-талассемию. Выявлены три такие мутации, одна из них характерна для уроженцев Средиземноморья (β39С—Т). Обнаружена рестриктаза (Mael), узнающая
4. Действие генов 91
этот сайт, благодаря чему стала возможной пренатальная диагностика β39-талассемии [1328а]. Делеции и инсерции, длина которых не кратна трем нуклеотидам, нарушают нормальное считывание генетической информации, в результате чего синтезируются нефункциональные цепи глобина. В различных популяциях обнаружено семь подобных мутаций, вызывающих β°-талассемию (табл. 4.17). Мутации, нарушающие процессинг РНК. Процессинг РНК заключается в вырезании интронов и сплайсинге (сращивании) экзонов с образованием функциональной мРНК (рис. 4.40, 4.52). Описано много различных мутаций, нарушающих этот процесс. Например, известна целая группа мутаций, изменяющих динуклеотид GT (или AG) в донорном (или акцепторном) участке сплайсинга. Эти динуклеотиды входят в состав так называемых канонических (обобщенных) последовательностей, которые включают еще несколько нуклеотидов и играют центральную роль в нормальном сплайсинге. В результате одиночных замен в этих сайтах сплайсинг нарушается, что приводит к β°- или β+-талассемии. В некоторых случаях мутации активируют так называемые криптические сайты с динуклеотидами GT и AG. В норме, вероятно, эти сайты в сплайсинге не участвуют. В результате образование нормальной мРНК нарушается. Мутации в интронах могут генерировать новые сайты сплайсинга в интронах, которые конкурируют с нормальными сайтами, замедляя процессинг, и в конечном счете приводят к талассемии. Мутации другого класса усиливают уже существующие криптические сайты. Часто встречающаяся мутация такого типа НbЕ активирует такой сайт. Это приводит к ослаблению синтеза гемоглобина HbβE и, следовательно, к талассемии. Различные мутации, нарушающие сплайсинг, перечислены в табл. 4.17. Все точковые мутации в гене β-глобина, которые вызывают β-талассемию, показаны на рис. 4.53. Делеции в β -глобиновом кластере генов и наследственная персистенция фетального гемоглобина. В отличие от а-талассемии Р-талассемия обычно обусловлена не делениями генов. Однако из этого правила есть много исключений. Более трети случаев Р-талассемии у индийцев оказывается связанной с делецией длиной 619 п. н., которая начинается во втором интроне и заканчивается за 3'-концом некодирующей области гена Нbβ (рис. 4.54, табл. 4.18). Различные редкие делеции в этом гене описаны у негров США, известен один случай среди датчан. Обнаружено также несколько других, более крупных делеции в γ-δ-β-локусе. Их локализация и протяженность показаны на рис. 4.54. Методами цитогенетики эти делеции обнаружить не удается: они слишком малы для микроскопического изучения. Мутации «гемоглобин Lepore» и «гемоглобин Kenya» представляют собой делеции, при которых сохраняются части функциональных генов и возникают их слияния: δ-β (гемоглобин Lepore) и γ-β (гемоглобин Kenya). Известно несколько делеции, при которых происходит утрата всего или почти всего кластера, при этом γ-, δ- и β-цепи не синтезируются. Принято различать делеции, приводящие к талассемическому фенотипу анемии, например δ-β-талассемия, и делеции, при которых отсутствие δ- и β-локусов компенсируется синтезом фетального гемоглобина (наследственная персистенция фетального гемоглобина, НПФГ). Это отличие не является абсолютным, поскольку при НПФГ компенсация благодаря синтезу γ-цепи не бывает полной. Остается
92 4. Действие генов
неясным, почему некоторые делеции активируют ген фетального гемоглобина. В настоящее время этот вопрос интенсивно изучается. Наследственная персистенция фетального гемоглобина может быть вызвана мутациями неделеционной природы. В случае греческой разновидности НПФГ обнаружена точковая мутация в 5'-фланкирующей области Аγ гена в положении —117 [1040]. Другая точковая мутация в положении 202 5'-фланкирующей области гена HbGγ найдена у негров с неделеционной НПФГ [1093]. Считается, что последовательности, которые изменяются под действием этих мутаций, в норме играют ключевую роль в остановке синтеза у-цепи в постнатальный период. Изучение генетического контроля синтеза гемоглобина имеет большое значение для лечения талассемии и серповидноклеточной анемии, так как усиленный синтез гемоглобина HbF при этих заболеваниях мог бы обеспечить значительный терапевтический эффект. Гетероклеточная наследственная персистенция фетального гемоглобина [222]. Группа гетерогенных генетических состояний ха рактеризуется некоторым усилением синте за фетального гемоглобина (2-3%, иногд; более) и явно неравномерным распределе нием в популяции эритроцитов. Этим и
4. Действие генов 93
Дата добавления: 2015-12-16 | Просмотры: 1100 | Нарушение авторских прав |