|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Гипотеза «один ген – один фермент»
Первые исследования. После того как в 1902 г. Гэррод указал на связь генетического дефекта при алкаптонурии с неспособностью организма расщеплять гомогентизиновую кислоту, важно было выяснить специфический механизм, лежащий в основе этого нарушения. Поскольку тогда уже было известно, что метаболические реакции катализируются ферментами, можно было предположить, что именно нарушение какого-то фермента приводит к алкаптонурии. Такая гипотеза обсуждалась Дришем (в 1896 г.). Ее высказывали также Холдейн (1920 г., см. [1117]) и Гэррод (1923 г. [1091]). Важными этапами в развитии биохимической генетики стали работы Кюхна и Бутенандта [1178; 1027] по изучению окраски глаз у мельничной огневки Ephestia kuhniella и аналогичные исследования Бидла и Эфрусси на Drosophila (1936) [987]. В этих пионерских работах для выяснения механизмов действия генов были выбраны мутанты насекомых, изученные ранее генетическими методами. Однако такой подход не привел к успеху. Проблема оказалась слишком сложной, и чтобы решить ее, необходимо было: 1) подобрать простой модельный организм, удобный для экспериментального изучения; 2) искать генетическую основу биохимических признаков, а не биохимическую основу генетически детерминированных признаков. Оба условия были выполнены в работе Бидла и Татума в 1941 году [988] (см. также Бидл, 1945 [986]).
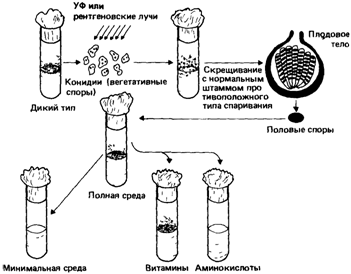
Модель Бидла и Татума. Статья этих исследователей начиналась так: «С точки зрения физиологической генетики - развитие и функционирование организма может быть сведено к сложной системе химических реакций, которые каким-то образом контролируются генами. Вполне логично предположить, что эти гены... либо сами выступают в роли ферментов, либо определяют их специфичность. Известно, что генетики-физиологи обычно пытаются исследовать физиологические и биохимические основы уже известных наследственных признаков. Этот подход позволил установить, что многие биохимические реакции контролируются специфическими генами. Такие исследования показали, что ферменты и гены обладают специфичностью одного порядка. Однако возможности этого подхода ограниченны. Наиболее серьезное ограничение заключается в том, что при этом в поле зрения исследователей попадают наследственные признаки, не имеющие летального эффекта и, следовательно, связанные с реакциями, которые не очень существенны для жизнедеятельности организма. Второе затруднение... заключается в том, что традиционный подход к проблеме подразумевает использование внешне проявляющихся признаков. Многие из них представляют собой морфологические вариации, основанные на системах биохимических реакций, настолько сложных, что их анализ необычайно затруднен. Подобные соображения привели нас к следующему выводу. Изучение общей проблемы генетического контроля биохимических реакций, определяющих развитие и метаболизм, должно проводиться с помощью процедуры, противоположной общепринятой: вместо того чтобы пытаться выяснить химические основы известных наследственных признаков, необходимо установить, обеспечивают ли гены контроль известных биохимических реакций и как они это делают. Нейроспора, относящаяся к аскомицетам, обладает свойствами, позволяющими реализовать такой подход и одновременно служит удобным объектом для генетических исследований. Вот почему наша программа была построена на использовании именно этого организма. Мы исходили из того, что облучение рентгеном вызывает мутации в генах, контролирующих определенные химические реакции. Пусть для выживания в данной среде организм должен осуществлять какую-то химическую реакцию, тогда мутант, лишенный такой способности, в этих условиях окажется нежизнеспособным. Однако его можно поддерживать и изучать, если выращивать в среде, к которой добавлен жизненно необходимый продукт генетически блокированной реакции». 4 Действие генов 9
Далее Бидл и Татум приводят описание схемы эксперимента (рис. 4.1). В состав полной среды входил агар, неорганические соли, солодовый экстракт, дрожжевой экстракт и глюкоза. Минимальная среда содержала только агар, соли, биотин и источник углерода. Наиболее подробно были исследованы мутанты, которые росли на полной среде и не росли на минимальной. Чтобы установить соединение, синтез которого нарушен у каждого из мутантов, в минимальный агар вносили отдельные компоненты полной среды. Таким способом были выделены штаммы, неспособные синтезировать определенные факторы роста: пиридоксин, тиамин и парааминобензойную кислоту. Было показано, что эти дефекты обусловлены мутациями в специфических локусах. Работа положила начало многочисленным исследованиям на нейроспоре, бактериях и дрожжах, в которых было установлено соответствие «генетических блоков», ответственных за отдельные метаболические этапы, и специфических нарушений ферментов. Этот подход очень быстро превратился в инструмент, позволяющий исследователям раскрывать метаболические пути. Гипотеза «один ген - один фермент» получила прочное экспериментальное подтверждение. Как показали работы последующих десятилетий, она оказалась удивительно плодотворной. Анализ дефектных ферментов и их нормальных вариантов позволил вскоре выявить такой класс генетических нарушений, которые приводили к изменению функции фермента, хотя сам белок по-прежнему обнаруживался и сохранял иммунологические свойства. В других случаях менялся температурный оптимум активности фермента. Некоторые варианты можно было объяснить мутацией, влияющей на общий регуляторный механизм и изменяющей в результате активность целой группы ферментов. Подобные исследования привели к созданию концепции регуляции активности генов у бактерий, которая включала и концепцию оперона. 10 4. Действие генов
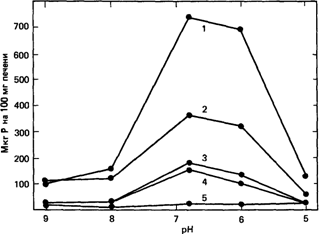
Первые примеры ферментативных нарушений у человека. Первым наследственным заболеванием человека, для которого удалось показать ферментативное нарушение, была метгемоглобинемия с рецессивным типом наследования (Гибсон и Харрисон, 1947 [1100]; Гибсон, 1948 [1099]) (25080). В этом случае поврежденным ферментом является NADH - зависимая метгемоглобин-редуктаза. Первая попытка систематического изучения группы заболеваний человека, связанных с дефектами метаболизма, была предпринята в 1951 году. При исследовании болезни накопления гликогена [1044] супруги Кори показали, что в восьми из десяти случаев патологического состояния, которое диагностировалось как болезнь Гирке (23220), структура гликогена печени представляла собой нормальный вариант, а в двух случаях была явно нарушена. Было также очевидно, что гликоген печени, накапливаясь в избытке, не может быть непосредственно превращен в сахар, поскольку у больных проявляется тенденция к гипогликемии. Для расщепления гликогена с образованием глюкозы в печени необходимы многие ферменты. Два из них-амило-1,6-глюкозидаза и глюкозо6-фосфатаза-были выбраны для изучения как возможные дефектные элементы ферментной системы. В гомогенатах печени при различных значениях рН было измерено освобождение фосфата из глюкозо-6фосфата. Результаты представлены на рис. 4.2. В нормальной печени обнаруживалась высокая активность с оптимумом при рН 6-7. Сильное нарушение функции печени при циррозе коррелировало с незначительным уменьшением активности. С другой стороны, в случае болезни Гирке с летальным исходом, активность фермента обнаружить вообще не удалось; такой же результат был получен при обследовании второго подобного больного. У двух пациентов с менее выраженными симптомами наблюдалось значительное уменьшение активности. Было сделано заключение, что в указанных случаях болезни Гирке с летальным исходом имел место дефект глюкозо-6-фосфатазы. Однако в большинстве более легких случаев активность этого фермента оказалась не ниже, чем при циррозе печени, и только у двух больных она была несколько меньшей (рис. 4.2). По мнению супругов Кори, аномальное накопление гликогена в мышечной ткани нельзя связывать с недостатком глюкозо-6-фосфатазы, поскольку в мышцах этот фермент отсутствует и в норме. В качестве возможного объяснения гликогеноза мышц они предположили нарушение активности амило-1,6-глюкозидазы. Это предсказание вскоре подтвердилось: Форбс [1081] обнаружил такой дефект при одном из клинически выраженных случаев болезни накопления гликогена с вовлечением сердечной и скелетных мышц. Сейчас нам
4. Действие генов 11
известно большое число ферментативных дефектов при болезни накопления гликогена [1133, 1244]. Хотя по степени проявления различные формы этого заболевания несколько различаются, в клиническом отношении между ними много общего. За одним исключением, все они наследуются по аутосомнорецессивному типу. Если бы ферментативные дефекты не были раскрыты, патология накопления гликогена рассматривалась бы как одно заболевание с характерными внутрисемейными корреляциями по тяжести течения, деталям симптоматики и срокам летального исхода. Таким образом, перед нами пример, когда генетическая гетерогенность, которую можно было лишь предполагать на основании изучения фенотипа (разд. 3.3.5), подтвердилась при анализе на биохимическом уровне: исследование ферментативной активности позволило идентифицировать специфические гены. В последующие годы темп исследований в области ферментативных дефектов нарастал, и для 588 идентифицированных рецессивных аутосомных генов, которые Мак-Кьюсик описывает в шестом издании своей книги «Менделевское наследование у человека» (1983) [133], более чем в 170 случаях обнаружены специфические ферментативные нарушения. Наши успехи в этой области непосредственно связаны с развитием концепций и методов молекулярной генетики. Некоторые этапы изучения ферментативных нарушений у человека. Мы приводим лишь наиболее важные вехи этого продолжающегося процесса: 1934 Фёллинг открыл фенилкетонурию [1080] 1941 Бидл и Татум сформулировали гипотезу «один ген - один фермент» [988] 1948 Гибсон описал первый случай ферментативного нарушения при заболевании у человека (рецессивная метгемоглобинемия) [1099] 1952 Супруги Кори обнаружили недостаточность глюкозо-6-фосфатазы при болезни Гирке [1044] 1953 Джервис продемонстрировал отсутствие фенилаланингидроксилазы при фенилкетонурии [1144]. Бикель сообщил о первой попытке смягчить ферментативное нарушение, применив диету с низким содержанием фенилаланина [1004] 1955 Смитис разработал методику электрофореза в крахмальном геле [1307, 1308] 1956 Карсон и др. обнаружили дефект глюкозо-6-фосфат— дегидрогеназы (G6PD) в случае индуцированной гемолитической анемии [1030] 1957 Калькар и др. описали ферментативную недостаточность при галактоземии, показав, что у человека и бактерий наблюдается идентичное нарушение ферментативной активности [1150] 1961 Крут и Вайнберг продемонстрировали дефект фермента при галактоземии in vitro в культуре фибробластов [1177] 1967 Сигмиллер и др. обнаружили дефект гипоксантин-гуанин—фосфорибозилтрансферазы (HPRT) при синдроме Леша —Найхана [1295] 1968 Кливер описал нарушение эксцизионной репарации при пигментной ксеродерме [1035] 1970 Нейфельд выявил ферментативные дефекты при мукополисахаридозах, что позволило идентифицировать пути расщепления мукополисахаридов [1240] 1974 Браун и Голдстейн доказали, что генетически детерминированная суперпродукция гидроксиметилглютарилСоА-редуктазы при семейной гиперхолестеринемии обусловлена дефектом локализованного в мембране рецептора липопротеинов низкой плотности, который модулирует активность этого фермента (HMG) [1023] 1977 Слай и др. продемонстрировали, что маннозо-6-фосфат (как компонент лизосомальных ферментов) узнается рецепторами фибробластов. Генетический дефект процессинга препятствует связыванию лизосомных ферментов, в результате нарушается их выход в цитоплазму и последующая секреция в плазму (I-клеточная болезнь) 12 4. Действие генов
1980 При псевдогипопаратиреозе обнаружен дефект белка, обеспечивающего сопряжение рецептора и циклазы. Дата добавления: 2015-12-16 | Просмотры: 1854 | Нарушение авторских прав |