|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
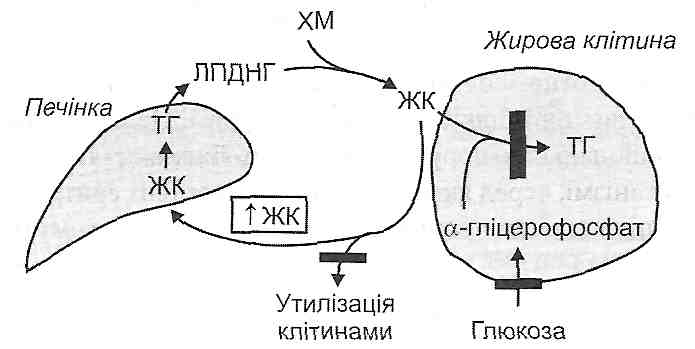
Додаткові ознаки клінічної смерті 13 страница5. Схуднення. При нелікованому цукровому діабеті порушується здатність жирової тканини перетворювати вільні жирові кислоти плазми крові в тригліцериди. Це пов'язано з гальмуванням ліпогенезу при відсутності інсуліну й пригніченням реакцій гліколізу, які необхідні для цього процесу (рис. 68).
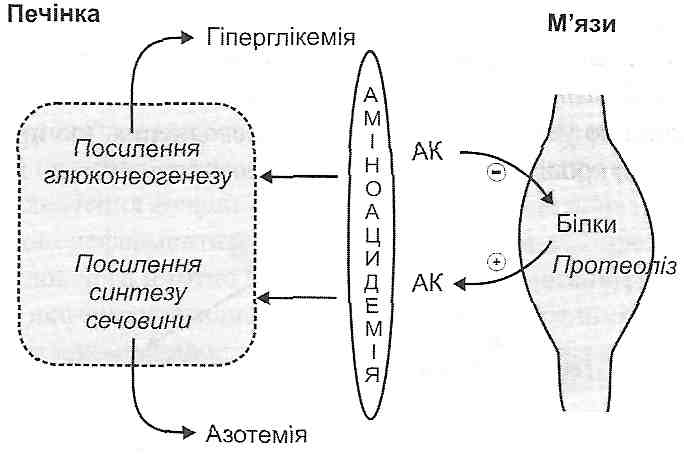
Рис. 68. Порушення депонування жирових кислот (ЖК) у жировій тканині при цукровому діабеті: ТГ— тригліцериди; ХМ — хіломікрони; ЛПДНГ '— ліпопротеїди дуже низької густини Посилення ліполізу під дією контрінсулярних гормонів також сприяє зменшенню маси жирової клітковини. 6. Атеросклероз (див. запит. 20.33). 20.30. Чим виявляють себе порушення білкового обміну при цукровому діабеті? 1. Аміноацидемією — збільшенням вмісту амінокислот у плазмі крові (рис. 69). В основі цього лежить зменшення транспорту амінокислот у м'язові клітини (при відсутності інсуліну зменшується проникність клітинних мембран для амінокислот) і посилення протеолізу в м'язах, унаслідок чого вивільнені амінокислоти надходять у кров. Надлишок вільних амінокислот поглинається печінкою, де посилюються процеси їх перетворення в глюкозу (глюконеогенез). Це, в кінцевому підсумку, призводить до подальшого збільшення рівня гіперглікемії.
Рис. 69. Порушення обміну білків при цукровому діабеті: АК—амінокислоти інсуліну. Клінічно пригнічення білоксинтетичних процесів виявляє себе порушеннями фізичного й розумового розвитку дітей, уповільненням загоєння ран, порушеннями утворення антитіл, унаслідок чого збільшується чутливість до інфекцій, часто розвивається фурункульоз.
20.31. Які порушення водно-електролітного обміну характерні для цукрового діабету? Який їхпатогенез?
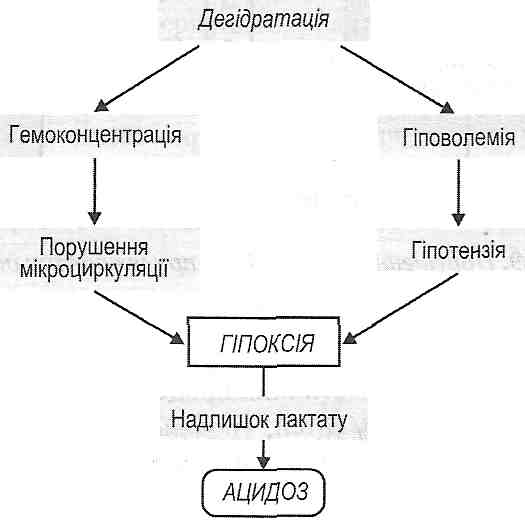
Рис. 70. Механізми дегідратації при цукровому діабеті 2. Гіперкаліємія. Є наслідком активації внутрішньоклітинного протеолізу. Відбувається вивільнення зв'язаного з білками калію, і його іони виходять з клітин у тканинну рідину і кров. 3. Гіпонатріємія. Якщо процеси ацидогене-зу в дистальних звивистих канальцях ниркових нефронів не забезпечують повного відтитровування гідрокарбонатного буфера, то якась частина іонів натрію втрачається із сечею разом з аніонами органічних кислот (ацетооцтової, р-оксимасля-ної). 20.32. Які порушення кислотно-основного стану розвиваються при цукровому діабеті? Для цукрового діабету характерний негазовий ацидоз. Залежно від механізмів його розвитку виділяють: а) кетонемічний метаболічний ацидоз - пов'язаний з накопиченням кетонових тіл; б) лактацидемічний метаболічний ацидоз — пов'язаний з накопиченням молочної кислоти. Причиною утворення останньої є зневоднення, що призводить до гіпо-волемії, згущення крові (гемоконцентрації) і, як наслідок, — до гіпоксії (рис. 71).
Рис. 71. Механізми лактацидеміїпри цукровому діабеті 20.33. Які варіанти коматозних станів можуть розвиватися при цукровому діабеті? 1. Діабетична кетонемічна кома. В основі її розвитку лежать ацидоз та інтоксикація, обумовлені кетоновими тілами. 2. Гіперосмолярна кома. Розвивається внаслідок дегідратації головного мозку, обумовленої високим ступенем гіперглікемії (див. запит. 20.26). 3. Лактацидемічна кома. Обумовлена накопиченням молочної кислоти й пов'язаним із цим ацидозом. 4. Гіпоглікемічна кома. Може розвиватися в результаті передозування інсуліну при лікуванні цукрового діабету. 84.Ускладнення цукрового діабету. Причини та механізми різних видів ком при цукровому діабеті. Віддалені ускладнення діабету. Які ускладнення характерні для цукрового діабету?... Макроангюпатії, мікроангюпатії, нейропатії. 20.35. Які механізми можуть лежати в основі розвитку макроангіопатій при цукровому діабеті? Макроангіопшпії характеризуються прискореним розвитком атеросклерозу в артеріях хворих на цукровий діабет. Найчастіше вражаються вінцеві артерії серця, артерії головного мозку й нижніх кінцівок. Це може призводити до розвитку таких ускладнень, як інфаркт міокарда, інсульт, гангрена пальців ніг і всієї стопи. Існують дві концепції, що пояснюють патогенез макроангіопатій. I. Концепція порушеного гомеостазу (власне діабетична). Головне значення в розвитку атеросклерозу при цукровому діабеті надається загальним порушенням обміну речовин в організмі, а саме: гіперглікемії, гіперліпопротеїнемії й ацидозу. Патогенетичне значення гіперглікемії полягає в тому, що вона: 1) є причиною неферментативного глікозилювання ліпопротеїдів плазми крові, унаслідок чого істотно збільшується їхня атерогенність; 2) викликає неферментативне глікозилювання мембранних білків ендотеліаль-них клітин і, як наслідок, призводить до підвищення проникності судинної стінки; 3) активує сорбітоловий шлях перетворення глюкози в гладких м'язових клітинах судин. Останнє відбувається, якщо концентрація глюкози в крові перевищує 20 ммоль/л. Результатом активації сорбітолоеого шляху є утворення в клітинах фруктози., Оскільки плазматична мембрана непроникна для цієї речовини, вона накопичується в цитоплазмі, підвищуючи осмотичний тиск внутрішньоклітинної рідини, викликаючи набряк і ушкодження клітин. Гіперліпопротеїнемія при цукровому діабеті характеризується збільшенням вмісту в крові ліпопротеїдів дуже низької густини (ЛПДНГ) і появою "модифікованих" ліпопротеїдів (ЛП): глікозильованих і ацетоацетильованих ЛІХ Про значення цих порушень у розвитку атеросклерозу див. розд. 28.З виникненням ацидозу пов'язане підвищення проникності судинної стінки й ушкодження її гладком'язових і ендотеліальних клітин - фактори, що сприяють атеросклерозу. II. Інсулінова концепція. її прихильники вважають, що провідною ланкою в патогенезі діабетичних макроангіопатій є гіперінсулінемія. Збільшення вмісту інсуліну в крові може бути ендогенним, як при цукровому діабеті II типу, і екзогенним, як результат передозувань інсуліну при лікуванні діабету І типу. Інсулін у великих кількостях, маючи мітогенну дію, викликає проліферацію гладком'язових клітин артеріальної стінки, що призводить до формування фіброзних атеросклеротичних бляшок. 20.36. Як пояснюють розвиток мікроангіопатій при цукровому діабеті? Чим вони можуть виявляти себе? Мікроангіопатїї— це ураження судин мікроциркуляторного русла (артеріол, капілярів), що виникають як ускладнення цукрового діабету. Сутність цих уражень полягає в значному збільшенні товщини базальної мембрани мікросудин, що утруднює обмін речовин між кров'ю й тканинами. Серед механізмів розвитку мікроангіопатій велике значення мають збільшення синтезу глікопротеїнів базальної мембрани і неферментативне глікозилювання її компонентів.Мікроангіопатії найчастіше виявляють себе ураженням судин нирок (діабетич- на нефропатія) і сітківки очей (діабетичнаретинопатія). Як наслідок, можуть розвиватися хронічна ниркова недостатність, відшарування сітківки. 20.37. Який патогенез нейропатій при цукровому діабеті? Heuponamu — це специфічні ураження нервових провідників у хворих на цукровий діабет. Вони виявляють себе розладами чутливості, вегетативних і рухових функцій, нервової трофіки. В основі патогенезу діабетичних нейропатій лежать процеси демієлінізації нервів і порушення аксоплазматичного транспорту. Суть демієлінізації полягає в руйнуванні мієлінової оболонки нервових волокон і порушенні утворення мієліну. Ці розлади пов'язують із: а) активацією сорбітолового шляху перетворення глюкози у шваннівських клітинах, що спричиняє їхнє ушкодження й загибель; б) пригніченням міоінозитолового шляху, внаслідок чого порушується утворення мі-оінозитолу - речовини, необхідної для побудови мієліну. 85.Експериментальне моделювання цукрового діабету. Принципи профілактики і терапії його основних типів. Профілактика ускладнень цукрового діабету. 20.38. Назвіть основні патогенетичні принципи лікування цукрового діабету. 1. Уведення інсуліну при інсулінозалежному цукровому діабеті І типу. Перспективними в цьому плані є трансплантація (3-клітин острівців підшлункової залози й застосування автоматизованих систем дозування і введення інсуліну. 2. Уведення фармакологічних препаратів, що усувають гіперглікемію, - гіпоглікемічних засобів (бігуаніди, похідні сульфонілсечовини). 3. Дієтотерапія, що забороняє вживати харчові продукти з високим вмістом цукру, регулює енергетичну цінність їжі та режим її споживання. 4. Фізичні навантаження (тренування). Вони зменшують рівень гіперглікемії й збільшують чутливість м'язової тканини до інсуліну. 86.Порушення ліпідного обміну: причини, механізми, прояви. Залежність розвитку дисліпопротеїнемій від факторів середовища, спадковості, супутніх захворювань. Принципи класифікації дисліпопротеїнемій. Етіологія і патогенез первинних (спадкових) і вторинних гіперліпопротеїнемій. Назвіть основні причини порушень жирового обміну в організмі. Причинами розладів жирового обміну можуть бути порушення: 1) перетравлювання і всмоктування ліпідів у тонкій кишці; 2) транспорту ліпідів кров'ю; 3) депонування ліпідів у жировій тканині; 4) жирової функції печінки (див. розд. 31); 5) проміжного обміну ліпідів у периферичних тканинах; 6) нервової й гормональної регуляції жирового обміну. 27.2. Що може бути причиною порушень перетравлювання і всмоктування ліпідів у кишках? 1. Порушення емульгування жирів: а) недостатнє надходження жовчі в кишки (механічна жовтяниця); б) передчасне руйнування жовчних кислот бактеріальною флорою при порушенні моторної функції кишок. 2. Порушення гідролітичного розщеплення жирів: а) недостатнє надходження панкреатичної ліпази у дванадцятипалу кишку; б) порушення активації цього ферменту за умов недостатньої секреції жовчі. 3. Порушення утворення ліпідних міцел у порожнині тонкої кишки: а) недостатнє надходження жовчі в тонку кишку; б) зв'язування жовчних кислот деякими лікарськими препаратами (холестира-мін, неоміцин та ін.); в) утворення кальцієвих солей жирових кислот при надмірному надходженні кальцію з їжею й водою. 4. Порушення всмоктування міцел: а) швидка евакуація вмісту тонкої кишки (проноси); б) ушкодження епітелію слизової оболонки тонкої кишки (ентерити, радіаційні ураження). 5. Порушення ресинтезу тригліцеридів і формування хіломікронів в епітеліальних клітинах кишок: а) зменшення кількості або пригнічення активності відповідних ферментів; б) дефіцит АТФ. 21.3. Які зміни складу крові можуть бути проявом порушень транспорту ліпідів в організмі? Гіперліпопротеїнемія і гіполіпопротеїнемія — відповідно збільшення й зменшення вмісту ліпопротеїдів у плазмі крові; дисліпопротеїнемія — порушення співвідно- шення між окремими класами ліпопротеїдів плазми крові; гіперліпацидемія — збільшення вмісту вільних жирових кислот у крові. 21.4. Які існують класи ліпопротеїдів плазми крові? У плазмі крові містяться хіломікрони (ХМ), ліпопротеїди дуже низької густини (ЛПДНГ), ліпопротеїди проміжної густини (ЛППГ), ліпопротеїди низької густини (ЛПНГ), ліпопротеїди високої густини (ЛПВГ). 21.5. Дайте порівняльну характеристику різних класів ліпопротеїдів плазми крові.
21.6. Як класифікують гіперліпопротеїнемії? За походженням гіперліпопротеїнемії бувають первинними (спадковими) і вторинними (набутими). Класифікація ВООЗ передбачає поділ гіперліпопротеїнемій на типи залежно від того, вміст ліпопротеїдів якого класу збільшений у крові (див. запит. 21.9). Залежно від механізмів розвитку гіперліпопротеїнемія може бути продукційною і ретенційною. 21.7. Які генетичні дефекти можуть бути причиною розвитку первинних (спадкових) гіперліпопротеїнемій? 1. Генетично обумовлені зміни структури апопротеїнів — білкової частини ліпопро-теїдних міцел, унаслідок чого ліпопротеїди плазми крові не можуть взаємодіяти з відповідними рецепторами або зазнавати ферментативних перетворень. 2. Спадкові дефекти ферментів, що беруть участь в обміні ліпопротеїдів, зокрема, дефіцит ліпопротеїнліпази, печінкової ліпази, лецитинхолестеролацилтрансфера-зи (ЛХАТ). 3. Аномалії клітинних рецепторів до ліпопротеїдів. 21.8. Назвіть можливі причини розвитку вторинних (набутих). гіперліпопротеїнємій. 1. Ендокринні хвороби (цукровий діабет, гіпотиреоз). 2. Метаболічні розлади (ожиріння, подагра). 3. Хвороби нирок (нефротичний синдром). 4. Хвороби печінки (обтураційна жовтяниця). 5. Інтоксикації (алкоголізм). 21.9. Наведіть класифікацію гіперліпопротеїнємій, запропоновану експертами ВООЗ.
Умовні позначення див. запит. 21.4. 21.10. Наведіть приклади розвитку різних типів гіперліпопротеїнємій за класифікацією ВООЗ.
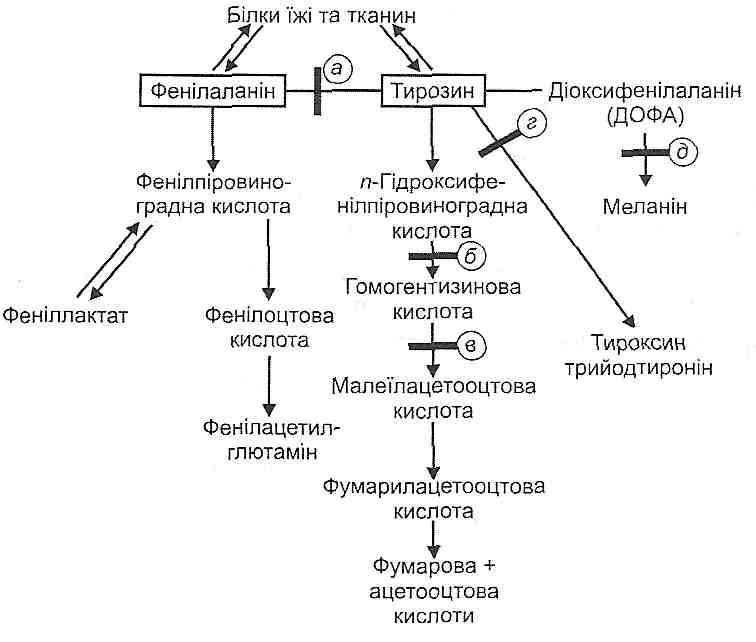
21.11. Що таке продукційна і ретенційна гіперліпопротеїнемія? Продукційна гіперліпопротеїнемія розвивається внаслідок збільшення утворення ліпопротеїдів, а ретенційна — у результаті порушення їх утилізації. 21.12. У чому полягає патогенетичне значення гіперліпопротеїнемій? Гіперліпопротеїнемії сприяють розвитку атеросклерозу (див. розд. 28). 21.13. Наведіть приклади гіполіпопротеїнемій. Дайте коротку характеристику. Зменшення вмісту в крові ліпопротеїдів (гіполіпопротеїнемія) спостерігається значно рідше, ніж гіперліпопротеїнемія. Причиною гіполіпопротеїнемій найчастіше є спадково обумовлені порушення. Серед них: 1) абеталіпопротеїнемїя. В організмі нема апопротеїну В, унаслідок чого не утворюються хіломікрони і ЛПНГ. Клінічно виявляє себе стеатореєю (поява жиру в калі) і авітамінозами жиророзчинних вітамінів. Успадковується аутосомно-рецесивно; 2) гіпобеталіпопротеїнемія. Зменшено вміст ЛПНГ. Вважають, що це доброякісний стан, який сприяє довголіттю, оскільки перешкоджає розвитку атеросклерозу й ішемічної хвороби серця. Успадковується аутосомно-домінантно; 3) танжерська хвороба (від назви острова Танжер на східному узбережжі США). Характеризується повною відсутністю ЛПВГ або наявністю їх аномальних форм. Порушується транспорт холестеролу від тканин у печінку, він накопичується в периферичних клітинах. Клінічно це виявляє себе гепатомегалією, спленомегалією, збільшенням лімфатичних вузлів. 21.14. Що таке "модифіковані" ліпопротеїди? Наведіть приклади. Яке їх патогенетичне значення? "Модифікованими" називають якісно змінені ліпопротеїди (ЛП). До них, зокрема, відносять глікозильовані ЛП (зв'язані з глікозильними групами, утворюються при гіперглікемії), ацетоацетильовані ЛП (зв'язані з ацетооцтовою кислотою, утворюються при цукровому діабеті); ЛП, зв'язані з продуктами пероксидного окиснення ліпідів; ліпопротеїн X (з'являється при обтураційній жовтяниці); комплекси ліпопро-теїд-антитіло. "Модифіковані" ЛП є атерогенними. їх накопичення сприяє розвитку атеросклерозу. 87.Ожиріння: визначення поняття, класифікації; етіологія і патогенез окремих форм. Експериментальне моделювання ожиріння. Медичні проблеми, пов'язані з ожирінням. Що таке первинне і вторинне ожиріння? Первинним називають ожиріння, що являє собою самостійний патологічний процес. Вторинне ожиріння є ознакою тих чи тих захворювань. Найпоширенішими варіантами вторинного ожиріння є церебральне і гормональне ожиріння. 21.16. Назвіть основні причини первинного ожиріння. 1. Надмірне споживання їжі, що перевищує енергетичні витрати організму. 2. Гіподинамія — обмеження фізичної активності людини. 3. Генетична схильність. Може виявляти себе особливостями харчової поведінки людини або особливостями регуляції жирового обміну. 21.17. У яких випадках виникає церебральне ожиріння? Церебральне ожиріння виникає при ураженнях гіпоталамуса, де зосереджені центри, що регулюють харчову поведінку. Такі ураження виникають при травмах, пухлинах, енцефаліті. Провідним механізмом ожиріння в цьому випадку є поліфагія (підвищення апетиту). 21.18. Які гормональні порушення можуть бути причиною вторинного ожиріння? Гормональне ожиріння розвивається як одна з ознак ендокринних хвороб. Воно супроводжує розвиток: а) гіпотиреозу; б) аденоми острівців підшлункової залози (гіперінсул інізм); в) синдрому Іценка—Кушинга; г) гіпофункції статевих залоз. 21.19. Чим відрізняється гіперпластичний тип ожиріння від гіпертрофічного? Гіперпластичне ожиріння пов'язане з гіперплазією жирових клітин, тобто зі збільшенням їхньої кількості. Для нього характерними є початок у ранньому дитячому віці і велика надлишкова вага. В основі гіпертрофічного ожиріння лежить збільшення маси окремих жирових клітин, при цьому їхня кількість не міняється. Ожиріння цього типу має більш пізній початок і не настільки виражене, як у попередньому випадку. 21.20. Які існують експериментальні моделі ожиріння? I. Експериментальні моделі первинного ожиріння. Отримано чисті лінії мишей і щурів з генетично обумовленим ожирінням, що є ознакою, яка передається від потомства до потомства. II. Експериментальні моделі вторинного церебрального ожиріння: а) руйнування вентромедіальних ядер гіпоталамуса, що утворюють "центр насичення"; б) електростимуляція латеральних ядер гіпоталамуса, що становлять "центр апетиту". III. Експериментальні моделі вторинного гормонального ожиріння: а) вимикання функції деяких ендокринних залоз (видалення щитоподібної залози, кастрація); б) введення в організм великої кількості деяких гормонів (інсуліну, глюкокор-тикоїдів). IV. Експериментальні моделі місцевого ожиріння — перетинання симпатичних нервів. При цьому в тканині з порушеною іннервацією збільшується маса жирової клітковини, оскільки припиняється ліполітична дія катехоламінів. 21.21. Які механізми лежать в основі збільшення маси жирової тканини при ожирінні? I. Посилення ліпогенезу. Цей механізм є провідним при: а) посиленому надходженні жирових кислот у жирові клітини з хіломікронів і ліпопротеїдів дуже низької густини (переїдання, церебральне ожиріння, гіперінсулінізм); б) посиленому утворенні жирових кислот в адипоцитах із глюкози (надмірне споживання вуглеводів, гіперінсулінізм, гіперфункція кори надниркових залоз); в) збільшенні активності ферментів ліпогенезу (гіперінсулінізм). II. Пригнічення ліполізу. В основі цього механізму зменшення активності гормончут-ливої ліпази адипоцитів. Це буває при: а) гіподинамії; б) гіпотиреозі; в) порушенні симпатичної іннервації. 21.22. У чому полягає патогенетичне значення ожиріння? При ожирінні значно зростає ризик виникнення багатьох соматичних хвороб. Серед них — атеросклероз, ішемічна хвороба серця, інфаркт міокарда, цукровий діабет II типу. 88.Позитивний і негативний баланс азоту. Види гіперазотемії. Зміни білкового складу крові. Спадкові порушення обміну амінокислот. У дорослої здорової людини кількість азотистих речовин, що виводяться з організму, дорівнює кількості, що він її отримує з їжею. Такий стан називається азопшстою рівновагою. В організмі, що росте, при вагітності, при введенні або надмірному утворенні! анаболічних гормонів, при посиленому годуванні після виснажливих хвороб азоту виводиться менше, ніж його надходить. У цьому випадку йдеться про позитивний азотистий баланс. І навпаки, якщо азоту виводиться більше, ніж надходить, то розвивається негативний азотистий баланс. Це може бути при голодуванні, втраті білків через нирки (протеїнурія), шкіру (опіки), кишки (проноси); при тиреотоксикозі, інфекційній гарячці 22.2. Назвіть основні причини аліментарної білкової недостатності. Аліментарнії білкова недостатність розвивається внаслідок порушень надходження в організм білків, їх перетравлювання і всмоктування. Основними її причинами є голодування, незбалансоване за амінокислотним і складом харчування, запальні й дистрофічні зміни різних відділів кишок, що супроводжуються порушеннями їх секреторної та моторної функцій. 22.3. Назвіть основні причини порушення біосинтезу білків у клітинах. 1. Порушення структури генів, що кодують інформацію про будову білків (мутації). 2. Отрути й специфічні інгібітори мультиферментних комплексів, що забезпечують ] процеси транскрипції, трансляції й посттрансляційної модифікації білків. 3. Дефіцит незамінних амінокислот. 4. Дефіцит АТФ. 5. Порушення утворення транспортних і рибосомної РНК, білків рибосом. 22.4. Порушення яких етапів можуть спричиняти розлади біосинтезу білків у клітинах? I. Порушення транскрипції- утворення інформаційної РНК на матриці ДНК. II. Порушення трансляції— синтезу поліпептидних ланцюгів з амінокислот. III. Порушення посттрансляційної модифікації білків — формування їхньої третин-; ної й четвертинної структур. 22.5. Які зміни білкового складу крові можуть виникати в умовах патології? I. Гіпопротеїнемія - зменшення вмісту білків у плазмі крові. Виникає головним чином за рахунок зниження кількості альбумінів і може бути набутою і спадковою. До гіпопротеїнемії спричиняються голодування, аліментарна білкова недостатність, захворювання печінки, вихід білків із кровоносного русла (крововтрата, плазмовтрата, ексудація, транссудація) і втрата білків із сечею (протеїнурія). Гіпопротеїнемія веде до зменшення онкотичного тиску плазми крові, у результаті чого рідина виходить з кровоносних судин в інтерстиціальну тканину - розвиваються набряки. II. Гіперпротеїнемія — збільшення вмісту білків у плазмі крові. Буває відносною (згущення крові) і абсолютною. Абсолютна гіперпротеїнемія найчастіше обумовлена збільшенням синтезу білків плазми крові і головним чином у-глобулінів (антитіл). Клінічні прояви гіперпротеїнемії пов'язані зі збільшенням в'язкості крові, зміною її реологічних властивостей і, як наслідок, порушеннями мікроциркуляції. III. Диспротеїнемія - зміна співвідношення між окремими білковими фракціями крові. Може бути спадковою і набутою. Часто пов'язана зі зміною спектру а- і у-глобулінів. Характерна для гострих запальних процесів ("білки гострої фази запалення"), дифузних захворювань сполучної тканини, аутоімунних захворювань. Іноді в крові з'являються якісно змінені білки, зокрема, парапротеїни і кріогло-буліни. Парапротеїни — це імуноглобуліни, що є продуктами одиничних клонів лімфоцитів. їх поява обумовлена проліферацією окремих антитілосинтезуючих клітин, що буває при патологічних процесах пухлинної природи (мієломна хвороба, макроглобулінемія Вальденстрема). Кріоглобуліни — це різновид парапротеїнів. Вони являють собою патологічні білки із властивостями імуноглобулінів, що преципітують при охолодженні. 22.6. Що таке продукційна іретенційна гіперазотемія? Пперазотемія — це збільшення залишкового (небілкового) азоту в крові. У нормі залишковий азот на 50 % складається з азоту сечовини, близько 25 % його припадає на частку амінокислот, інша частина — на інші азотисті продукти. Продукційна (печінкова) гіперазотемія виникає внаслідок порушення утворення сечовини, детоксикації азотистих продуктів у печінці. Ретенційна (ниркова) гіперазотемія є наслідком порушення видільної функції нирок. 22.7. Які фактори можуть викликати порушення утворення сечовини в печінці? Порушення синтезу сечовини може спостерігатися на завершальних етапах розвитку недостатності печінки як одна з ознак порушення її детоксикаційної функції. Можливі спадково обумовлені порушення сечовиноутворення. їх причиною може бути недостатній синтез цілого ряду ферментів: аргінінсукцинатліази (аргінін- сукцинатурія), карбамоїлфосфатсинтетази і орнітинкарбамоїлтрансферази (амоніє-мія) і аргінінсукцинатсинтетази (цитрулінурія). Наслідком порушення синтезу сечовини є накопичення аміаку в крові й цілий ряд пов'язаних з цим тяжких клінічних проявів (докладно див. розд. 31). 22.8. У чому сутність і чим виявляють себе спадково обумовлені порушення обміну фенілаланіну? Спадково обумовленим порушенням обміну фенілаланіну є фенілкетонурія -за-хворювання з аутосомно-рецесивним типом спадкування. Його причиною є генетичний дефект ферменту фенілаланінгідроксилази, що в нормі перетворює фенілаланін на тирозин (рис. 73). За відсутності зазначеного ферменту окиснення фенілаланіну відбувається шляхом утворення фенілпіровиноградної і фенілмолочної кислот. Однак цей шлях має малу пропускну здатність, і тому фенілаланін накопичується у великій кількості в крові, тканинах і спинномозковій рідині, що в перші ж місяці життя веде до важкого ураження центральної нервової системи й невиліковного слабоумства. Рис. 73. Блокада шляхів метаболізму фенілаланіну і тирозину: блок а - фенілкетонурія; б — тирозиноз; в — алкаптонурія; г - гіпотиреоз; д — альбінізм 22.9. У чому сутність і чим виявляють себе спадково обумовлені порушення обміну тирозину? Залежно від рівня генетичних дефектів спадково обумовлені порушення обміну тирозину можуть виявлятися розвитком тирозинозу, алкаптонурії, альбінізму. Усі ці захворювання успадковуються аутосомно-рецесивно. Тирозиноз виникає внаслідок генетичного дефекту ферменту - оксидази парагі-дроксифенілпіровиноградної кислоти. У результаті вона, будучи першим проміжним продуктом обміну тирозину, не перетворюється в гомогентизинову кислоту, накопичується в крові й разом з тирозином виводиться із сечею. Алкаптонурія є наслідком порушення синтезу оксидази гомогентизинової кислоти, що перетворює останню в малеїлацетооцтову кислоту. У результаті в крові й сечі з'являється гомогентизинова кислота. Сеча при стоянні на повітрі, а також при додаванні до неї лугу стає чорною, що пояснюється окисненням гомогентизинової кислоти киснем повітря й утворенням алкаптону. Гомогентизинова кислота з крові проникає в тканини — хрящову, сухожилля, зв'язки, внутрішній шар стінки аорти, унаслідок чого з'являються темні плями в ділянці вух, носа, на склерах. Іноді розвиваються тяжкі зміни в суглобах. Альбінізм обумовлений дефіцитом ферменту тирозинази. Унаслідок цього не утворюється пігмент шкіри й волосся — меланін. Організм, позбавлений пігменту, стає дуже чутливим до дії ультрафіолетового випромінювання. Дата добавления: 2016-03-26 | Просмотры: 420 | Нарушение авторских прав |
 2. Порушеннями біосинтезу білків. Це прямо пов'язано з випадінням анаболічної дії
2. Порушеннями біосинтезу білків. Це прямо пов'язано з випадінням анаболічної дії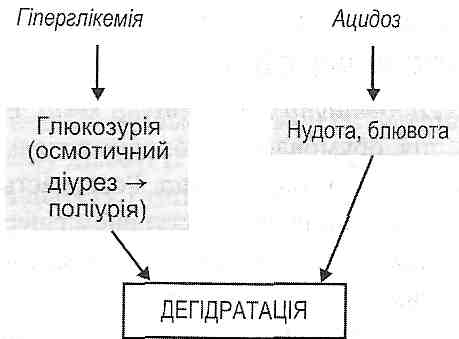 1. Зневоднення (дегідратація). Є наслідком поліурії. Посилює дегідратацію блювота, яка часто супроводжує ацидоз, що розвивається у хворих на цукровий діабет (рис. 70).
1. Зневоднення (дегідратація). Є наслідком поліурії. Посилює дегідратацію блювота, яка часто супроводжує ацидоз, що розвивається у хворих на цукровий діабет (рис. 70).