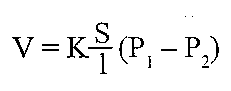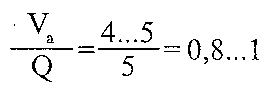|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Артеріальна гіпертензія, обумовлена прийомом ліків28.24. Які існують експериментальні моделі артеріальної гіпертензії? Жодна хвороба людини не має такої великої кількості різних експериментальних моделей, як артеріальна гіпертензія. Нині артеріальну гіпертензію вивчають на мишах, щурах, кролях, кішках, собаках, свинях, мавпах. За методами відтворення всі моделі артеріальної гіпертензії можна поділити на кілька великих груп. I. Порушення функції центральної нервової системи: а) зіткнення процесів умовного збудження і гальмування, що призводить до розвитку у тварин (собак, мавп) неврозу; б) моделювання психоемоційної напруги шляхом створення зоосоціального конфлікту (у мавп), змін біоритмів, іммобілізації тварин; в) електрична і хімічна стимуляція лімбічних структур головного мозку. II. Порушення мозкового крово- і лімфообігу: а) одно- і двостороннє перев'язування сонних і вертебральних артерій, що живлять мозок (центрально-ішемічна артеріальна гіпертензія)', б) блокада лімфовідведення по периневральних і периваскулярних лімфатичних шляхах за допомогою каоліну, що його вводять у велику цистерну мозку. III. Порушення функції депресорних регуляторних систем: а) двостороннє перетинання у кролів і собак депресорних і синусних нервів, у результаті чого знімаються гальмівні впливи з барорецепторів рефлексогенних зон дуги аорти і каротидного синуса (рефлексогенна гіпертензія, або гіпертензія розгальмування); б) центральна деаферентація барорецепторів, що її викликають ушкодженням ядра солітарного тракту; в) пригнічення синтезу простагландинів за допомогою індометацину. IV. Порушення функції нирок: а) звужування обох ниркових артерій або звужування однієї ниркової артерії з видаленням другої контрлатеральної нирки (реноваскулярна гіпертензія). Виникнення артеріальної гіпертензії в цьому випадку пов'язане з активацією ренін-ангіотензинної системи; б) видалення обох нирок і переведення тварин на гемодіаліз для запобігання уремії (ренопривна гіпертензія). її розвиток пояснюють припиненням депресорних функцій нирок; в) обгортання нирок целофаном, шовком. При цьому виникає перинефрит:' здавлюється ниркова паренхіма, розвивається венозний застій і гіпоксія нирок, активується ренін-ангіотензинна система. V. Порушення гормонального стану: а) введення тваринам адреналіну; в) введення вазопресину; в) субтотальне видалення кори надниркових залоз. При цьому відбувається посилення регенерації залозистої тканини з посиленою продукцією кортикостероїдів, особливо альдостерону (надшрнико-регенераційна гіпертензія). VI. Порушення водно-сольового обміну: а) введення тваринам великої кількості кухонної солі (сольова гіпертензія)', б) введення мінералокортикоїдів (дезоксикортикостерону, альдостерону) - мі-нералокортикоїдна гіпертензія; в) поєднане введення кухонної солі й мінералокортикоїдів. VII. Моделі генетично обумовленої артеріальної гіпертензії. У багатьох лабораторіях світу виведено чисті лінії щурів, характерною рисою яких є гіпертензія -ознака, що спадкується. Це, зокрема, щури зі спонтанною гіпертензією (лінія Окамото - Аокі); щури, схильні до інсультів; новозеландські щури, міланські щури; щури, чутливі до сольової дієти, та ін. 119. Первинна артеріальна гіпертензія як мультифакторіальне захворювання; сучасні уявлення про етіологію та патогенез гіпертонічної хвороби. Роль нирок в патогенезі первинної артеріальної гіпертензії. Артеріальна гіпертензія (АГ) - патологічний стан, який характеризується підвищенням артеріального тиску. Усі випадки гіпертонічної хвороби підрозділяються на гіпертонічну хворобу (ГХ, есенціальну гіпертензію) і симптоматичні АГ, у тому числі ізольовану (систолічну) АГ. Термін «гіпертонічна хвороба» вживається тоді, коли етіологія підвищення АТ не з’ясована. Якщо підвищення АТ є проявом якогось конкретного захворювання, воно трактується як симптоматична (вторинна) АГ. Найчастіше це спостерігається при ураженні нирок (пієлонефрит, гломерулонефрит), ендокринної системи (феохромоцитома, хвороба Іценка - Кушинга, синдром Конна та ін), природжених або набутих ураженнях судин (коарктація дуги аорти, звуження судин нирок), нервової системи (пухлини, травми головного мозку). Етіологія і патогенез. Основним етіологічним фактором ГХ вважають нервовопсихічне перенавантаження ЦНС, спричинене короткочасними гострими або тривалими нервовими негативними впливами. Внаслідок цього виникає активація генетичних дефектів людини. До них належать: порушення мембран клітин; скупчення іонів Са++; активність симпатичної нервової системи; ренінангіотензивна система; рецептори до ангіотензину ІІ; гіпертрофія серця. Всі ці явища регулюються генами і деякою мірою зовнішніми факторами. Внаслідок цього зростають серцевий викид, загальний периферичний судинний опір, розвивається АГ. У свою чергу, АГ спричинює шемію мозку, серця, судин, нирок, надниркових залоз. Експериментально та клінічно доведено, що в цих органах відбувається збільшення вмісту тканинного ангіотензину. Існують різні шляхи утворення ангіотензину ІІ, а не тільки за участю ангіотензинперетворюючого ферменту (АПФ). Циркулюючий ангіотензин ІІ підвищує загальний периферичний судинний опір, що збільшує АТ, а тканинний ангіотензин ІІ збільшує ішемію органів. Тканинний ангіотензин ІІ спричинює гіпертонічну енцефалопатію, порушення мозкового кровообігу, гіпертрофію і фіброз міокарда, гіпертрофію артерій м’язового типу, артеріосклероз, гломерулосклероз, підвищення рівнів катехоламінів і альдостерону. Все це сполучене з добовою динамікою АТ. Таким чином, ГХ - це полігенний дефект, який поки що не можна подолати. Вилікувати хворих на ГХ неможливо, але активно запобігти її тяжким органним ускладненням - це реально. Відповідно до останніх рекомендацій ВООЗ та Міжнародного товариства гіпертензії (1999) виділяють декілька рівнів АТ. Цю класифікацію рекомендується застосовувати для з’ясування стадії як гіпертонічної хвороби (есенціальної гіпертензії), так і вторинної гіпертензії. Якщо виявлено ГХ, діагноз формулюється з визначенням її стадії та характеру ураження органівмшеней (серце, головний мозок, очне дно, нирки, судини). Перебіг захворювання відображають стадії підвищення АТ і стан органівмішеней. На початку захворювання самопочуття хворих може бути цілком задовільним або проявлятися у вигляді специфічних церебральних і кардіальних скарг: головний біль у потиличній, тім’яній, скроневій ділянках; запаморочення; шум у голові, вухах; минущі порушення зору; дратівливість; безсоння; колючі болі у ділянці серця, серцебиття, задишка. У міру прогресування хвороби виникають об’єктивні ознаки порушення функцій органівмішеней і різноманітні ускладнення. Перебіг ГХ часто ускладнюється гіпертонічними кризами. Криз - це гостре погіршання стану хворого, що призводить до значного і відносно короткочасного підвищення АТ. Існують 2 типи кризів. Для кризів І типу характерні нейровегетативні зрушення, які супроводжуються гіперсимпатикотонією, що призводить до різкого збільшення викиду крові за одиницю часу і підвищення загального периферичного судинного опору. Для кризів ІІ типу характерними є підвищення у крові альдостерону, затримка натрію в організмі, що спричинює збільшення об’єму крові. Гіпертонічний криз І типу супроводжується різким підвищенням АТ, головним чином систолічного, інтенсивним головним болем, запамороченням, пітливістю, тремором, тахікардією, іноді порушенням зору, нудотою, блювотою. Продовжується нетривалий час (від кількох годин до 1 доби) і може закінчитися urina spastica. Розвиток кризів ІІ типу поступовий, протягом декількох діб, при цьому підвищується як систолічний, так і діастолічний АТ. Хворі на вигляд часто бліді, пригнічені, сонливі; з одутлим обличчям; спостерігаються набряки рук і ніг; у літніх хворих знижене сприйняття кольору. 120. Гіпертензія малого кола кровообігу (первинна, вторинна). Причини та механізми розвитку. Клінічні та гемодинамічні прояви. 29.20. Які причини і механізми розвитку гіпертензії малого кола кровообігу? Гіпертензія малого кола кровообігу характеризується збільшенням тиску в легеневій артерії понад 25 мм рт. ст. її розвиток може бути обумовлений такими механізмами: а) тривалий спазм артеріол легень. Найчастіше виникає в результаті зменшення парціального тиску кисню в альвеолярному повітрі, що буває при гіпоксичній гіпоксії (див. розд. 19) і порушеннях вентиляції легень; б) гострий рефлекторний спазм легеневих артеріол. Розвивається при емболії судин легень, стенозі отвору двостулкового клапана. В останньому випадку вмикається рефлекс Китаєва: збільшення тиску в лівому передсерді і легеневих венах викликає збудження барорецепторів і спазм легеневих артеріол, що попереджає збільшення гідростатичного тиску в капілярах легень і розвиток набряку; в) збільшення тиску повітря в бронхах і альвеолах. Викликає здавлення легеневих капілярів і, як наслідок, збільшення судинного опору в малому колі кровообігу. Буває у людей під час важких нападів кашлю. При цьому тиск у легеневій артерії може зростати до 250 мм рт. ст.; г) облітерація легеневих судин (артеріол, капілярів, венул) унаслідок ураження їхніх стінок (наприклад, при емфіземі легень). В експерименті показано, що гіпертензія малого кола кровообігу виникає при вимиканні не менше 2/3 судинного русла. Отже, видалення однієї легені не призводить до розвитку цього синдрому; ґ) збільшення хвилинного об'єму серця більше ніж у 3 рази; д) порушення відтоку крові по легеневих венозних судинах (вади двостулкового клапана серця, недостатність лівого шлуночка, здавлювання легеневих вен); є) збільшення в 'язкості крові (наприклад, при поліцитемії); є) уроджені вади, пов'язані зі скиданням крові зліва направо (незарощення Боталло-вої протоки, дефекти міжшлуночкової перегородки). Залежно від того, на якій ділянці легеневих судин збільшується опір, розрізняють пре- і посткапілярну форму гіпертензії малого кола кровообігу. 29.21. Які механізми можуть лежати в основі розвитку набряку легень? Вихід рідини з кровоносних судин в інтерстиціальну тканину легень і альвеоли може бути обумовлений такими механізмами. 1. Гідростатичний механізм - різке збільшення гідростатичного тиску в капілярах легень. Набряк розвивається, коли гідростатичний тиск стає вищим за 30 мм рт. ст. (у нормі 6-9 мм рт. ст.), тобто його величина стає більшою за онкотичний тиск крові. Така ситуація може виникати при гострій лівошлуночковій недостатності серця, обумовленій великим інфарктом міокарда, при стенозі отвору двостулкового клапана (кардіогенний механізм), при введенні великих кількостей (кілька літрів) крово- і плазмозамінних розчинів хворим з порушеним діурезом (гіперво-лемічний механізм). 2. Мембраногенний механізм — збільшення проникності легеневих капілярів. Буває при: а) екзогенній інтоксикації (отруєння фосфорорганічними сполуками, наприклад, фосгеном); б) ендогенній інтоксикації (уремія, печінкова недостатність); в) алергічних реакціях І типу. 3. Онкотичний механізм — зменшення онкотичного тиску плазми крові. Відносно часто буває у хворих з нефротичними синдромом (див. розд. 32). З урахуванням патогенезу розрізняють дві фази розвитку набряку легень. I. Інтерстиціальний набряк - накопичення набрякової рідини в інтерстиціальній тканині легень. Клінічно виявляється нападами серцевої астми. Розвивається паренхіматозна недостатність дихання з явищами гіпоксемії. II. Альвеолярний набряк — перехід набрякової рідини в альвеоли. При цьому порушується їх вентиляція — розвивається вентиляційна недостатність дихання з явищами гіпоксемії і гіперкапнії. 29.22. Якими порушеннями виявляє себе синдром емболії малого кола кровообігу? Поява емболів у судинах легень викликає розвиток таких змін: 1) генеролізованш спазм артеріол усього малого кола кровообігу (а не тільки судин, де містяться емболи). Це викликає різку гіпертензію малого кола і розвиток гострої правопшуночкової недостатності серця (синдром гострого легеневого серця); 2) зменшення артеріального тиску у великому колі кровообігу. Пов'язане зі зменшенням хвилинного об'єму серця і зниженням тонусу артеріол у периферичних відділах (рефлекс Швічки—Паріна); 3) порушення зовнішнього дихання — розвиток паренхіматозної дихальної недостатності (див. запит. 29.17).
121. Артеріальна гіпотензія. Етіологія та патогенез гострих і хронічних артеріальних гіпотензій. 28.29. Як класифікують артеріальну гіпотензію? Які гемодинамічні фактори можуть лежати в основі її розвитку? Артеріальна гіпотензія (стійке зниження артеріального тиску) спостерігається частіше в осіб астенічної конституції і виявляється загальною адинамією, швидкою стомлюваністю, тахікардією, задишкою, запамороченням, головним болем, непритом-ностями і депресивним станом з періодичним підвищенням нервової збудливості. Артеріальну гіпотензію класифікують у такий спосіб. I. Фізіологічна (не супроводжується хворобливими симптомами). II. Патологічна (з характерним симптомокомплексом): 1. Гостра. 2. Хронічна: а) симптоматична (вторинна); б) нейроциркуляторна дистонія гіпотензивного типу (первинна). З огляду на те, що рівень артеріального тиску визначається величиною серцевого виштовху, об'ємом циркулюючої крові і тонусом резистивних судин, можливі три гемодинамічні форми артеріальної гіпотензії: 1) пов'язана з недостатністю скорочувальної функції серця; 2) викликана зменшенням об'єму циркулюючої крові; 3) така, що виникає внаслідок зниження тонусу резистивних судин. 28.30. Які причини і механізми розвитку хронічної артеріальної гіпотензії? Симптоматична (вторинна) хронічна артеріальна гіпотензія є наслідком низки загальних соматичних гострих і хронічних захворювань серця (вади, міокардит, інфаркт міокарда), головного мозку (комоція), легень (крупозна пневмонія), печінки (гепатит, механічна жовтяниця), крові (анемія), ендокринних залоз, а також екзогенних інтоксикацій. Стосовно нейроциркуляторної (первинної) артеріальної гіпотензії вважають, що її основним етіологічним і патогенетичним фактором, як і гіпертонічної хвороби, є перенапруження основних процесів кори великого мозку (збудження і гальмування). Однак, на відміну від первинної гіпертензії, спостерігається переважання гальмування і поширення його на підкіркові вегетативні утворення, зокрема на судиноруховий центр. 28.31. Які загальні і місцеві розлади гемодинаміки можуть бути пов'язані з первинними порушеннями функції ємнісних судин? До ємнісних судин належать вени, що депонують кров з метою її розподілу і повернення до серця. Було відзначено, що в судинах ділянки низького тиску міститься 70-80 % загального об'єму крові. Про важливе функціональне значення венозного відділу свідчить хоча б той факт, що одномоментне зменшення його ємності всього лише на 3 % подвоює венозне повернення до серця, а при однакових за величиною змінах тиску в артеріальній і венозній системах об'єм останньої змінюється приблизно в ЗО разів більше, ніж артеріальної. З урахуванням сказаного було зроблено висновок про те, що вже незначні порушення функції з боку ємнісних судин можуть призводити до істотних порушень загальної гемодинаміки. Такі розлади можуть виявлятися двома типами змін: а) розвитком артеріальної гіпертензії при збільшенні тонусу гладких м'язів венозних судин, унаслідок чого збільшується діастолічний приплив крові до серця; б) виникненням артеріальної гіпотензії— гострої (колапсу) або хронічної — при швидкому або тривалому збільшенні ємності венозної системи. Місцеві розлади кровообігу, пов'язані з порушенням функції вен, призводять до розвитку венозної гіперемії (дав. розд. 13).
122. Артеріосклероз: визначення поняття, класифікація. Характеристика основних форм: атеросклероз (Маршана), медіакальциноз (Менкеберга), артеріолосклероз. Артеріосклероз являє собою комбінацію чотирьох процесів: інфільтрації, проліферації, дегенерації і склерозування. Різні поєднання цих процесів у різних судинах визначають "мозаїчний" характер артеріосклеротичних уражень. 1. Інфільтрація - проникнення із плазми крові в судинну стінку і відкладення в ній ліпідів, складних вуглеводів і білків. 2. Проліферація — розмноження гладком'язових клітин артеріальної стінки, у результаті чого формуються так звані фіброзні "бляшки", що виступають у просвіт артерій і порушують течію крові в них. 3. Дегенерація— цим терміном позначають ушкодження і загибель клітин судинної стінки, а також розвиток дистрофічних змін, у тому числі кальцинозу. 4. Склерозування — посилене утворення сполучної тканини, що виявляється синтезом її основної інтерстиціальної речовини й волокнистих структур. 123. Атеросклероз. Етіологія атеросклерозу: фактори ризику, причинні фактори. Сучасні теорії атерогенезу - «запальна» і «рецепторна». Роль спадкових і набутих порушень рецептор-опосередкованого транспорту ліпопротеїнів в атерогенезі. Сучасне тлумачення атеросклерозу набагато ширше. За визначенням ВООЗ, атеросклероз — це різні поєднання змін інтими артерій, що виявляються у вигляді осередкового відкладення ліпідів, складних сполук вуглеводів, елементів крові і циркулюючих у ній продуктів, утворення сполучної тканини і відкладення кальцію. 28.5. Що таке артеріосклероз Менкеберга? Описана в 1903 р. Менкебергом на прикладі артерій нижніх кінцівок людини форма артеріосклерозу характеризується ураженням середньої оболонки (медії) артерій еластичного і еластично-м'язового типу і виявляється тріадою ознак: медіанекро-зом, медіакальцинозом і медіасклерозом. 28.6. Дайте порівняльну характеристику атеросклерозу й артеріосклерозу Менкеберга.
28.7. Чим виявляють себе склеротичні зміни кровоносних судин? Склеротично змінені судини вирізняються підвищеною щільністю й крихкістю. Унаслідок зниження еластичних властивостей вони не в змозі адекватно змінювати свій просвіт залежно від потреби органа або тканини у кровопостачанні. Спочатку функціональна неповноцінність склеротично змінених судин, а отже, органів і тканин виявляється тільки при підвищенні до них вимог, тобто при збільшенні навантаження. Подальше прогресування атеросклеротичного процесу може призвести до зниження працездатності і у стані спокою. Сильний ступінь атеросклеротичного процесу, як правило, супроводжується звуженням і навіть повним закриттям просвіту артерій. При повільному склерозу ванні артерій в органах з порушеним кровопостачанням відбуваються атрофічні зміни з поступовим заміщенням функціонально активної паренхіми сполучною тканиною. Швидке звуження або повне перекриття просвіту артерій часто веде до змертвіння ділянки органа з порушеним кровообігом, тобто до інфаркту. Інфаркт міокарда — найчастіше і найнебезпечніше ускладнення атеросклерозу вінцевих артерій. 28.8. Як в експерименті моделюють атеросклероз? У 1912 р. М. Анічков і С. Халатов запропонували спосіб моделювання атеросклерозу у кролів шляхом уведення всередину холестеролу (через зонд або домішуючи його до звичайного корму). Виражені атеросклеротичні зміни розвиваються через декілька місяців при щоденному використанні 0,5 г холестеролу на 1 кг маси тіла. Як правило, їх супроводжує підвищення рівня холестеролу в сироватці крові (у 3-5 разів, якщо порівнювати з вихідними величинами), що стало підставою для припущення про провідну патогенетичну роль у розвитку атеросклерозу гіперхолес-теролемії. Цю модель легко відтворюють не тільки у кролів, але і в курей, голубів, мавп, свиней. У собак і щурів, резистентних до дії холестеролу, атеросклероз відтворюють шляхом комбінованого впливу холестеролу і метилтіоурацилу, що пригнічує функцію щитоподібної залози. Таке поєднання двох факторів (екзогенного і ендогенного) веде до тривалої і вираженої гіперхолестеролемії. Додавання до їжі вершкового масла і солей жовчних кислот також сприяє розвитку атеросклерозу. У курей (півнів) експериментальний атеросклероз аорти розвивається після тривалої дії діетилстильбестролу. У цьому випадку атеросклеротичні зміни виявляються на тлі ендогенної гіперхолестеролемії, що виникає внаслідок порушення гормональної регуляції обміну речовин. 28.9. Що таке фактори ризику атеросклерозу? Що до них відносять? Факторами ризику атеросклерозу називають сукупність внутрішніх і зовнішніх умов, які в багато разів підвищують імовірність розвитку цього захворювання у людини. Проведені в багатьох країнах світу епідеміологічні дослідження дають можливість виділити цілий ряд факторів ризику атеросклерозу. 1. Вік. Різке збільшення частоти і тяжкості атеросклеротичних уражень судин у зв'язку з віком, особливо помітне після ЗО років, стало підставою для того, щоб деякі дослідники вважали атеросклероз функцією віку і винятково біологічною проблемою (І. Давидовський). Більшість учених, однак, дотримуються думки, що вікові і атеросклеротичні зміни судин — це різні форми артеріосклерозу, особливо на пізніх стадіях їхнього розвитку. При цьому вікові зміни судин сприяють розвитку атеросклербтичних уражень. Зазначений аспект проблеми атеросклерозу знайшов своє відображення в роботах М. М. Горева і очолюваної ним лабораторії Інституту геронтології НАН України. 2. Стать. У віці 40—70 років на атеросклероз та інфаркт міокарда атеросклеротичної природи чоловіки хворіють частіше, ніж жінки (у середньому в 3-4 рази). Після 70 років захворюваність на цю недугу серед чоловіків і жінок приблизно однакова. Зазначені відмінності, мабуть, пов'язані, з одного боку, з нижчим вихідним рівнем холестеролу і тим, що у жінок він міститься в сироватці крові в основному у фракції неатерогенних ліпопротеїдів високої густини, а з другого — з антисклеротичною дією жіночих статевих гормонів. 3. Спадковість. Роль спадкового фактора у виникненні атеросклерозу підтверджують статистичні дані про високу частоту ішемічної хвороби серця в окремих родинах, а також у однояйцевих близнюків. Мова йде про спадкові форми гіперліпо-протеїнемії і спадково обумовлені дефекти метаболізму артеріальної стінки. 4. Надлишкове харчування. Досвід країн з високим життєвим рівнем (США, Швеція, Чехія та ін.) переконливо доводить таку закономірність: що більше потреба в енергії задовольняється за рахунок тваринних жирів і продуктів, які містять хо-лестерол, то вищий вміст холестеролу в крові й відсоток захворюваності на атеросклероз. Навпаки, у країнах, де на частку жирів тваринного походження припадає незначна частина енергетичної цінності добового раціону (близько 10 %), захворюваність на атеросклероз низька (Японія, Китай). Існує також залежність між захворюваністю на атеросклероз і кількістю споживаного цукру. Якщо додати до цього, що 75-85 % хворих на цукровий діабет хворіють на атеросклероз і вмирають від нього, у 4/5 хворих на атеросклероз установлене зниження толерантності до глюкози, а 1/3 з них перебуває в предіабетичному стані, то певну роль у виникненні атеросклерозу варто відвести надмірному споживанню вуглеводів і порушенню їх утилізації. 5. Стрес. Є спостереження, які свідчать про те, що захворюваність на атеросклероз вища серед людей "стресових професій", тобто професій, що вимагають тривалої і сильної нервової напруги (лікарі, учителі, викладачі, працівники управлінського апарату, льотчики та ін.). У цілому захворюваність на атеросклероз вища серед міського населення, якщо порівнювати з сільським. Це може пояснюватися тим, що в умовах великого міста людина частіше зазнає нейрогенних стресових впливів. 6. Гіподинамія. Малорухливий спосіб життя, різке зменшення фізичного навантаження (гіподинамія) - ще один важливий фактор атерогенезу. Про це, зокрема, свідчать менша захворюваність на атеросклероз серед працівників фізичної праці і більша- в осіб, робота яких пов'язана з розумовою працею; швидша нормалізація рівня холестеролу в сироватці крові, після надмірного його надходження ззовні, під дією фізичних навантажень. В експерименті виявлено виражені атеросклеротичні зміни в артеріях кролів після того як вони тривалий час перебували у спеціальних клітках, що значно обмежували рухову активність тварин. Особливу атерогенну небезпеку являє собою поєднання малорухливого способу життя і надлишкового харчування. 1. Інтоксикація. Вплив алкоголю, нікотину, інтоксикація бактеріального походження та інтоксикація, викликана різними хімічними речовинами (фториди, CO, H2S, свинець, бензол, сполуки ртуті), також є факторами, що сприяють розвитку атеросклерозу. Більшість наведених тут інтоксикацій супроводжувалася не тільки загальними порушеннями жирового обміну, властивими атеросклерозу, але й типовими дистрофічними та інфільтративно-проліферативними змінами в артеріальній стінці. 8. Артеріальна гіпертензія. Підвищений артеріальний тиск набуває значення фактора, що сприяє розвитку атеросклерозу в комбінації з іншими, особливо якщо він перевищує 160/90 мм рт. ст. Так, при однаковому рівні холестеролу захворюваність на інфаркт міокарда при гіпертензії в п'ять разів вища, ніж при нормальному артеріальному тиску. В експерименті на кролях, у їжу яких додавали холестерол, атеросклеротичні зміни розвиваються швидше і досягають більшого ступеня на тлі артеріальної гіпертензії. 9. Гормональні порушення, хвороби обміну речовин. У деяких випадках атеросклероз виникає на тлі попередніх гормональних порушень (цукровий діабет, мікседема, зниження функції статевих залоз) або хвороб обміну речовин (подагра, ожиріння, спадкові форми гіперліпопротеїнемії і гіперхолестеролемії). Про етіологічну роль гормональних розладів у розвитку атеросклерозу свідчать і досліди з експериментального відтворення цих порушень у тварин шляхом впливу на ендокринні залози. 28.10. Які існують концепції патогенезу атеросклерозу? Відомі нині теорії патогенезу атеросклерозу можна звести до двох, принципово різних концепцій, що відрізняються між собою відповіддю на питання: що первинне, а що вторинне при атеросклерозі, інакше кажучи, що є причиною, а що наслідком - ліпоїдоз внутрішньої оболонки артерій чи дегенеративно-проліферативні зміни останньої. Відповідно до уявлень R Вірхова і його послідовників, при атеросклерозі спочатку розвиваються дистрофічні зміни внутрішньої оболонки стінки артерій, а відкладення ліпідів і солей кальцію — явище вторинного порядку. Перевагою даної концепції є те, що вона спроможна пояснити розвиток спонтанного і експериментального атеросклерозу як у тих випадках, коли є порушення ліпідного обміну, так і в тих (що особливо важливо), коли їх немає. Першорядну роль автори цієї концепції відводять артеріальній стінці, тобто субстрату, який безпосередньо втягується в патологічний процес. На противагу цим поглядам ще відтоді, коли М. Анічков і С. Халатов провели перші свої експерименти, успішно розвивається концепція про роль у розвитку атеросклерозу загальних метаболічних порушень в організмі, що супроводжуються гі-перхолестеролемією і гіперліпопротеїнемією. З цих позицій атеросклероз — наслідок первинної дифузної інфільтрації ліпідів, зокрема холестеролу, у незмінену внутрішню оболонку артерій. Подальші зміни в судинній стінці (явища мукоїдного набухання, дистрофічні зміни волокнистих структур і клітинних елементів субендотеліаль-ного шару, продуктивні зміни) розвиваються у зв'язку з відкладенням у ній ліпідів, тобто є вторинними. 28.11. У чому сутність плазмової теорії патогенезу атеросклерозу? Відповідно до плазмової теорії, основу патогенезу атеросклерозу та інфільтрації судинної стінки зокрема, становлять зміни хімічного складу плазми крові, що виникають унаслідок загальних порушень ліпідного обміну в організмі. Пріоритет у становленні цієї теорії належить М. М. Ані-чкову та його послідовникам. Плазмова теорія у своєму розвитку пройшла два етапи. ^ Перший етап — холестероловий. Сутність теорії на цьому етапі її розвитку зводилася до положення: "без холестеро-лу немає атеросклерозу". Вважалося, що причиною виникнення інфільтративних змін артеріальної стінки (ліпоїдозу) є збільшення вмісту холестеролу в плазмі крові — гіперхолес-теролемія. Після того, як стало відомо, що транспорт ліпідів, у тому числі і холестеролу, здійснюється у складі лшопротеїдів, виявилося, що для розвитку атеросклерозу має значення не стільки гіперхолестеролемія, скільки кількісні і якісні зміни ліпо-~~ протеїдів плазми крові. Настав другий етап розвитку плазмової теорії —ліпопротеїновий. Основою його стало положення: "без атерогенних ліпопротеїдів немає атеросклерозу
124. Недостатність зовнішнього дихання: визначення поняття, принципи класифікації. Патогенез основних клінічних проявів. Задишка: види, причини, механізми розвитку. Хвороба гіалінових мембран. Характеристика.
Недостатність зовнішнього дихання — це патологічний стан, при якому система зовнішнього дихання не здатна забезпечити нормальний склад газів крові (газовий гомеостаз). 29.2. Як класифікують дихальну недостатність? I. За клінічним перебігом розрізняють гостру і хронічну недостатність дихання. Гостра недостатність розвивається протягом кількох днів, годин і навіть хвилин, її прикладом може бути асфіксія (див. запит. 29.16). Хронічна недостатність розвивається протягом тривалого часу і є наслідком захворювань бронхів і легень (хронічна пневмонія, пневмосклероз, емфізема легень та ін.). II. За вираженістю клінічних ознак недостатність дихання може бути компенсованою і декомпенсованою. При компенсованій недостатності газовий склад крові ще не змінений (спрацьовують компенсаторні захисні механізми); при декомпенсованій - газовий гомеостаз порушений. III. За патогенезом виділяють два різновиди: а) вентиляційну і б) паренхіматозну недостатність зовнішнього дихання. Недостатність зовнішнього дихання - патологічний стан, при якому система зовнішнього дихання не здатна забезпечити нормальний склад газів крові (газовий гомеостаз). • Дихальну недостатність класифікують: 1) За клінічним перебігом розрізняють: а) гостру і б) хронічну недостатність дихання. Гостра недостатність розвивається протягом декількох днів, годин і навіть хвилин (асфіксія). Хронічна недостатність розвивається протягом тривалого часу і є наслідком захворювання бронхів і легень (хронічна пневмонія, пневмосклероз, емфізема легень і ін). 2) За клінічними проявами недостатність дихання може бути: а) компенсованою і б) декомпенсованою. При компенсованій недостатності газовий склад крові не змінюється із-за включення компенсаторних захисних механізмів; при декомпенсованій - газовий гомеостаз порушується. 3) За патогенезом виділяють два різновиди: а) вентиляційну і б) паренхіматозну. • Вентиляційна недостатність дихання виникає внаслідок порушень обміну газів між атмосферним повітрям і альвеолами легень, тобто в результаті порушень легеневої вентиляції (гіповентиляції). Для цього виду дихальної недостатності характерні наступні порушення гомеостазу: 1) зменшення напруги кисню (рО2) в артеріальній крові - гіпоксемія; 2) збільшення напруги вуглекислого газу (рСО2) в артеріальній крові - гіперкапнія; 3) газовий ацидоз. Причини вентиляційної недостатності дихання. □ Позалегеневі причини - безпосередньо не пов’язані із порушеннями бронхів і легень. Ними можуть бути: 1) порушення функції дихального центру, внаслідок: а) прямого впливу на нього різних патогенних факторів чи б) рефлекторного впливу на хемо- або барорецептори; 2) порушення функції мотонейронів спинного мозку, які іннервують дихальні м’язи, при пухлинах в спинному мозку, сирінгомієлії, поліомієліті; 3) порушення функції нервово-м’язового апарату при: а) ушкодженні нервів, які іннервують дихальні м’язи (запалення, авітаміноз, травма), б) затруднені передачі до м’язів нервового імпульсу (міастенія, ботулізм, правець), в) порушенні функції самих дихальних м’язів (міозит, дистрофія). Велике значення в акті дихання приймає діафрагма. Порушення роботи діафрагми може привести до значних розладів дихання, що трапляється при ушкодженні n.frenicus. При клонічних судомах м’язів діафрагми з’являється гикавка; 4) порушення рухливості грудної клітки, що веде до обмеження розтягання легень і зменшення альвеолярної вентиляції, при: а) вроджених або набутих деформація ребер і хребетного стовпа, скостенінні реберних хрящів, б) зрощенні листків плеври, в) асциті, метеоризмі, великій тучності, г) різких больових відчуттях, які виникають під час дихання (міжреберна невралгія, міозит); 5) порушення цілісності грудної клітки і плевральної порожнини та попадання у неї атмосферного повітря (пневмоторакс). □ Легеневі причини - пов’язані із патологічними процесами в легенях і повітряноносних шляхах, зокрема: 1) порушення прохідності повітроносних шляхів (бронхіти, бронхіальна астма, злоякісні пухлини); 2) порушення еластичнких властивостей легеневої тканини (емфізема, пневмосклероз); 3) зменшення кількості функціонуючих альвеол (пневмонія, набряк легень, ателектаз, пневмоторакс). • Патогенетичні варіанти вентиляційної недостатності дихання включають в себе: 1) дисрегуляційну, 2) рестрикційну та 3) обструкційну недостатність
125. Дисрегуляторні порушення альвеолярної вентиляції. Причини і механізми патологічного дихання (порушення частоти, глибини, ритму). Патогенез періодичного дихання у дітей.
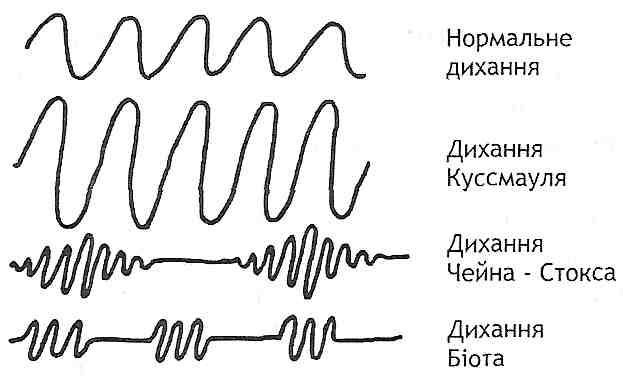
Дисрегуляційна недостатність виникає під впливом дії рефлекторних, гуморальних або інших чинників на дихальний центр можуть змінюватися ритм, глибина і частота дихання, а також може виникати задишка. Ці зміни можуть бути проявом компенсаторних реакцій організму, спрямованих на підтримку сталості газового складу крові, або проявом порушень нормальної регуляції дихання, що ведуть до розладів альвеолярної вентиляції, до недостатності дихання. Зміни центральної регуляції дихання можуть виявляти себе такими його типами. 1. Брадипное — рідке дихання. Механізм розвитку рідкого дихання полягає в зміні характеру нервової імпульсації, що йде від різних рецепторів до дихального центру, або в первинному порушенні діяльності самих дихальних нейронів. Рефлекторне зменшення частоти дихання може спостерігатися при підвищенні артеріального тиску (рефлекс із барорецепторів дуги аорти і каротидного синуса), при гіпероксії (унаслідок періодичного збудження хеморецепторів, чутливих до зниження напруги кисню в артеріальній крові). Глибоке рідке дихання може з'явитися при підвищенні опору руху повітря у верхніх дихальних шляхах — стенотичне дихання. У цьому випадку вдих і видих відбуваються повільніше, ніж звичайно. У встановленні такого типу дихання певну роль відіграють імпульси, що надходять у дихальний центр від міжреберних м'я-. зів, що працюють з підвищеним навантаженням. Крім того, має значення запізнювання в цьому випадку гальмівного рефлексу Герінга-Брейєра. Брадипное може розвиватися в результаті безпосередньої дії патогенних факторів на дихальний центр, що знижує збудливість дихальних нейронів. Пригнічення дихального центру можливе при тривалій і тяжкій гіпоксії (в умовах розрідженої атмосфери, при недостатності кровообігу та ін.), за умов впливу речовин з наркотичною дією, при деяких органічних ураженнях головного мозку (запалення, порушення мозкового кровообігу, набряк та ін.) і функціональних розладах центральної нервової системи (невроз, істеричні реакції). У всіх цих випадках рідке дихання може супроводжуватися зменшенням його глибини, що призводить до зниження альвеолярної вентиляції і розвитку недостатності дихання. 2. Поліпное (тахіпное) — часте поверхневе дихання. В основі розвитку поліпное лежить рефлекторна перебудова роботи дихального центру. У деяких тварин (наприклад, у собак) часте поверхневе дихання виникає при дії високої температури. У людини поліпное може спостерігатися при гарячці, функціональних порушеннях центральної нервової системи (істерія), ураженні легень (ателектаз, пневмонія, застійні явища). Крім того, до розвитку поліпное може спричинятися біль, що локалізується у ділянках тіла, які беруть участь у дихальному акті (грудна клітка, черевна стінка, плевра). Біль призводить до обмеження глибини дихання і збільшення його частоти (щадне дихання). Поліпное знижує ефективність дихання, тому що при цьому значно зменшується альвеолярна вентиляція і газообмін в основному відбувається у "мертвому" просторі. 3. Гіперппое — глибоке часте дихання. В умовах патології гіперпное розвивається при інтенсивній рефлекторній або гуморальній стимуляції дихального центру, наприклад, при зниженні парціального тиску кисню у вдихуваному повітрі або при підвищенні в ньому концентрації С02, при анемії, ацидозі і т. д. Крайній ступінь збудження дихального центру виявляється диханням Куссмауля, яке найчастіше спостерігається у хворих у стані діабетичної коми. Воно являє собою гучне часте дихання, при якому після глибокого вдиху настає посилений видих з активною участю експіраторних м'язів. 4. Апное— тимчасова зупинка дихання. Тимчасова зупинка дихання може бути пов'язана зі зменшенням рефлекторної або безпосередньої хімічної стимуляції дихального центру. Наприклад, апное виникає у тварини або людини після пасивної гіпервентиляції під наркозом унаслідок зменшення в артеріальній крові напруги CO і припиняється зразу ж, як тільки вміст С02 нормалізується. При швидкому підвищенні артеріального тиску, наприклад при введенні в кров адреналіну, також, спостерігається апное (рефлекс із барорецепторів). Апное, що часто повторюється і порушує звичайний ритм дихання, може бути пов'язане зі зниженням збудливості нейронів дихального центру (гіпоксія, інтоксикація, органічні ураження головного мозку). 29.7. Що таке періодичне дихання? Які відомі його форми? Періодичним диханням називають таке порушення ритму дихання, при якому періоди дихання чергуються з періодами апное. Найчастіше бувають два типи періодичного дихання: дихання Чейна-Стокса і дихання Біота. Дихання Чейна—Стокса характеризується наростанням амплітуди дихання до вираженого гіперпное, а потім зменшенням її до апное, після якого знову настає цикл дихальних рухів, що закінчуються також апное (рис. 135).
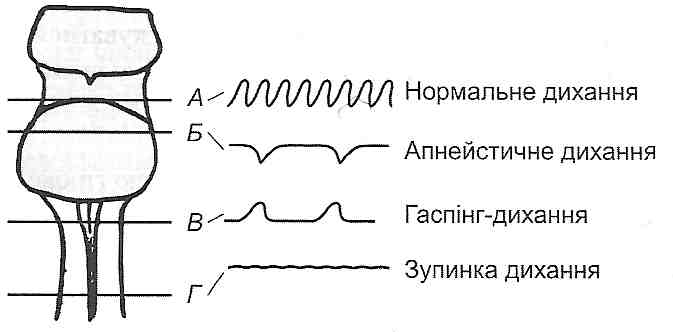
Рис. 135. Дихання Куссмауля і деякі види періодичного дихання Циклічні зміни дихання у людини можуть супроводжуватися потьмаренням свідомості в період апное і його нормалізацією в період збільшення вентиляції. Артеріальний тиск при цьому також коливається, як правило, підвищуючись у фазі посилення дихання і знижуючись у фазі його ослаблення. У більшості випадків дихання Чейна-Стокса є ознакою гіпоксії головного мозку. Воно може виникати при недостатності серця, захворюваннях мозку і його оболонок, уремії. Деякі лікарські препарати (наприклад, морфін) також можуть викликати дихання Чейна — Стокса. Його можна спостерігати і у здорових людей на великій висоті (особливо під час сну), у недоношених дітей, що, очевидно, пов'язане з недосконалістю нервових центрів. Патогенез дихання Чейна-Стокса можна представити в такий спосіб. Клітини кори великого мозку і підкіркових утворень унаслідок гіпоксії пригнічуються — дихання зупиняється, свідомість зникає, пригнічується діяльність судинорухового центру. Однак хеморецептори при цьому все ще здатні реагувати на зміни вмісту газів у крові. Різкого посилення імігульсації з хеморецепторів, поряд із прямою дією на центри високої концентрації вуглекислого газу і стимулами з барорецепторів унаслідок зниження артеріального тиску, виявляється достатньо для збудження дихального центру — дихання поновлюється. Поновлення дихання веде до оксигенації крові, що зменшує гіпоксію головного мозку і поліпшує функцію нейронів судинорухового центру. Дихання стає глибше, свідомість проясняється, підвищується артеріальний тиск, поліпшується наповнення серця. Вентиляція, що збільшується, веде до підвищення напруги кисню і зниження напруги С02 в артеріальній крові. Це у свою чергу призводить до ослаблення рефлекторної і хімічної стимуляції дихального центру, діяльність якого починає вгасати, - настає апное. Дихання Біота відрізняється від дихання Чейна-Стокса тим, що дихальні рухи, які характеризуються постійною амплітудою, раптово припиняються, так само як і раптово починаються. Найчастіше дихання Біота спостерігається при менінгіті, енцефаліті та інших захворюваннях, що супроводжуються ушкодженням центральної нервової системи, особливо довгастого мозку. 29.8. Що таке термінальне дихання? Чим воно характеризується? Термінальним називають дихання, що виникає в термінальних станах, тобто за умов, коли організм перебуває на межі життя і смерті. Найчастіше реєструють два типи термінального дихання: апнейстичне і гаспінг-дихання. Апнейстичие дихання характеризується конвульсивним зусиллям вдихнути, що не припиняється, але зрідка переривається видихом. В експерименті його спостерігають після перетинання у тварини обох блукаючих нервів і мозкового стовбура між пневмотаксичним (у ростральній частині мосту) і апнейстичним (у середній і каудальній частинах мосту) центрами (рис. 136). Вважають, що апнейстичний центр має здатність збуджувати інспіраторні нейрони, які періодично гальмуються імпульсами із блукаючого нерва і пневмотаксичного центру. Перетинання зазначених структур призводить до постійної інспіраторної активності апнейстичного центру.
Рис. 136. Розвиток термінального дихання після перетинання стовбура мозку нарізних його рівнях (А, Б, В, Г) Гаспінг-дихання - це поодинокі, рідкі вдихи, частота і амплітуда яких поступово зменшується аж до повної зупинки дихання. Такий вид дихання спостерігають при агонії, наприклад у заключній стадії асфіксії. Звичайно характерні для гаспінг-дихання рідкі і низькоамплітуцні вдихи виникають після тимчасової зупинки дихання (претермінальної паузи). Поява їх, можливо, пов'язана зі збудженням клітин, що містяться в каудальній частині довгастого мозку, після вимикання функції вище розташованих відділів мозку. 29.9. Що таке задишка? Які механізми її розвитку? Задишка (дисппое) - це відчуття нестачі повітря і пов'язана з цим потреба посилити дихання. Відчуваючи нестачу повітря, людина не тільки мимовільно, але й свідомо збільшує активність дихальних рухів, прагнучи позбутися цього тяжкого відчуття, наявність якого і є найбільш істотною відмінністю диспное від інших видів порушення регуляції дихання (гіперпное, поліпное та ін.). Тому у людини, що втратила свідомість, задишки не буває. Задишка виникає у тих випадках, коли впливи, що збуджують вдих, є сильнішими за впливи, що його пригнічують, а також у разі підвищення чутливості дихального центру до чинників, які стимулюють дихання. Найважливішими з цих впливів є такі. 1. Збудження рецепторів, що стимулюють центр вдиху, і які активуються при сильному зменшенні об'єму легеневих альвеол (сильнішому, ніж при максимальному видиху). При патології може виникнути постійна імпульсація від цих рецепторів. Наприклад, при застійних явищах у легенях (недостатність серця, пневмонія) переповнені кров'ю судини, що оточують альвеоли, здавлюють їх, ємність альвеол зменшується, що веде до збудження рецепторів спадіння. 2. Збудження рецепторів інтерстиціальної тканини легень (J-рецепторів). Усі патологічні процеси, що ведуть до застійних явищ у легенях (пневмонія, недостатність серця), можуть викликати тривале збудження J-рецепторів і підвищену стимуляцію дихальних нейронів. 3. Рефлекси з повітроносних шляхів. Можуть викликатися твердими частинками, парами хімічних сполук та іншими чинниками, що потрапили в повітроносні шляхи і подразнюють так звані іритантнірецептори (мають властивості одночасно ме-хано- і хеморецепторів), розташовані в субепітеліальному шарі трахеї, бронхів і бронхіол. Значне збудження іритантних рецепторів спостерігають при бронхітах, бронхопневмонії, бронхіальній астмі і хворобах, при яких у бронхах і альвеолах міститься слиз, ексудат або транссудат). 4. Рефлекси з барорецепторів аорти і сонної пазухи. Ці рефлекси долучаються до патогенезу задишки при крововтраті, шоку, колапсі. За умов зменшення артеріального тиску до рівня 70 мм рт. ст. і нижче, різко зменшується імпульсація від цих рецепторів, яка в нормі гальмує центр вдиху (через активацію центру видиху). \ 5. Рефлекси з хеморецепторів аорти і сонної пазухи. При зниженні в крові напруги 02, підвищенні напруги С02 або збільшенні концентрації іонів водню відбувається посилене збудження рецепторів, розташованих в аортальному і каротидному тільцях, і, як наслідок, — посилене збудження центру вдиху. Цей механізм відіграє важливу роль у розвитку задишки при ацидозі, недостатності дихання, при анемії і т. д. 6. Безпосередня стимуляція нейронів дихального центру. У довгастому мозку є хеморецептори, вибірково чутливі до вуглекислого газу. Сильне збудження цих рецепторів при гіперкапнії також сприяє розвитку задишки. 7. Рефлекси з дихальних м 'язів. Відчуття нестачі кисню може виникнути при надмірному розтягненні міжреберних м'язів і сильному збудженні рецепторів розтягу, імпульсація від яких надходить у вищі відділи головного мозку. Цей механізм діє під час виконання тяжкої фізичної роботи, що потребує значної роботи інспіра-торних м'язів, при зменшенні еластичності легень, звуженні верхніх дихальних шляхів. 8. Стимуляція дихального центру продуктами власного метаболізму. Ідеться про накопичення вуглекислого газу, кислих продуктів обміну і зниження напруги кисню безпосередньо в нервових центрах унаслідок порушення мозкового кровообігу (спазм або тромбоз судин головного мозку, набряк мозку, колапс). Дихання при задишці, як правило, часте і глибоке. Посилюється як вдих, так і видих, який за умов задишки носить активний характер і відбувається за участю експіраторних м'язів. Однак у деяких випадках може переважати або вдих, або видих, тоді ведуть мову про інспіраторну (посилений вдих) або експіраторну (посилений видих) задишку. Інспіраторну задишку спостерігають, наприклад, у першій стадії асфіксії, при загальному збудженні центральної нервової системи, при фізичному навантаженні у хворих з недостатністю кровообігу, при пневмотораксі. Експіраторна задишка буває рідше і виникає головним чином при бронхіальній астмі, емфіземі, коли при видиху збільшується опір потокові повітря в нижніх дихальних шляхах
126. Порушення альвеолярної вентиляції. Обструктивні та рестриктивні механізми розвитку.
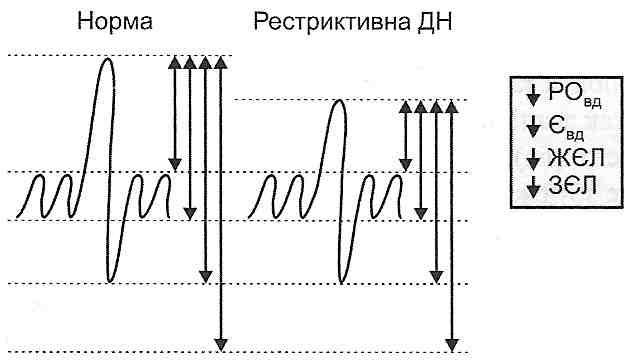
29.10. Що таке рестриктивна недостатність дихання? Які її причини? PecmpuKmuenoio називають дихальну недостатність, пов'язану з обмеженням зовнішнього дихання (restrict - обмеження; рис. 137).
Рис. 137. Зміни показників вентиляції легень при рестриктивній дихальній недостатності (ДН) Таке обмеження може бути обумовлене: 1) зменшенням дихальної поверхні легень, що буває після видалення сегмента, частки або цілої легені; при руйнуванні великих ділянок легень (туберкульоз); у результаті спадіння легеневої тканини (ателектаз, пневмоторакс); 2) збільшенням пружного опору легень — порушенням їхньої здатності розправлятися під час вдиху. Така ситуація закономірно виникає при: а) зменшенні розтяжності легень у зв'язку із заміщенням еластичних структур легеневої тканини колагеновими (пневмосклероз); б) збільшенні сили поверхневого натягу в альвеолах при порушеннях сурфак-танту (зменшенні утворення або посиленому руйнуванні).
29.13. Що таке обструктивна недостатність дихання? Коли вона розвивається? Обструктивною називають дихальну недостатність, яка виникає внаслідок збільшення аеродинамічного опору повітроносних шляхів. Основним фактором, що викликає таке збільшення, є зменшення радіуса повітроносних трубок (бронхів, бронхіол). Причинами обструктивної недостатності дихання є: 1) спазм гладких м 'язів бронхів (бронхіальна астма); 2) набряк слизових оболонок (бронхіти, бронхіоліти); м 3) здавлювання бронхіол (емфізема легень).
127. Бронхіальна астма. Етіологія: фактори ризику, причинні фактори. Патогенез. Клінічна картина. Особливості у дітей.
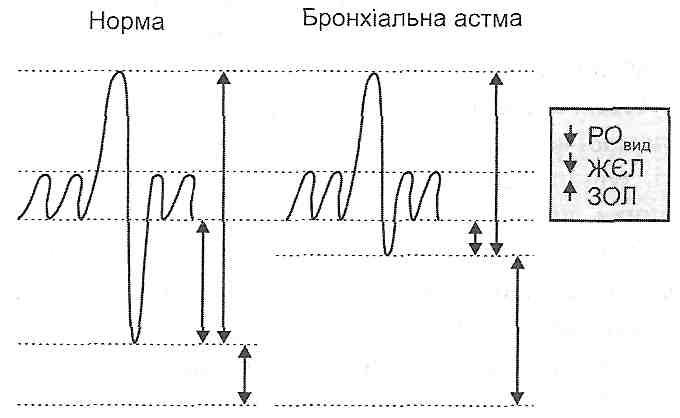
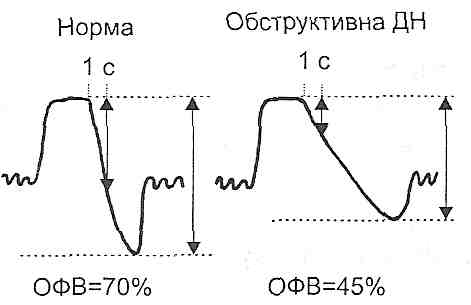
Бронхіальна астма — це алергічне захворювання, що характеризується повторними нападами експіраторної задишки, викликаної дифузним порушенням прохідності бронхів. Під час тривалих нападів бронхіальної астми розвивається обструктивна недостатність дихання. Для бронхіальної астми характерні такі зміни показників зовнішнього дихання (рис. 139): 1) зменшення резервного об'єму видиху; 2) зменшення життєвої ємності легень; 3) збільшення залишкового об'єму легень; 4) зменшення об'єму форсованого видиху (проба Тифно; рис. 140).
Рис. 139. Зміни показників вентиляції легень при бронхіальній астмі
Рис. 140. Об'єм форсованого видиху (ОФВ; тест Тифно) при обструктивній дихальній недостатності (ДН)
128. Причини і механізми порушень дифузії газів у легенях. Порушення легеневого кровообігу. Порушення загальних і регіональних вентиляційно-перфузійних взаємовідношень у легенях.
29.18. Назвіть причини порушення дифузії газів у легенях. Дифузія газів через альвеоло-капілярну мембрану здійснюється відповідно до першого закону Фіка:
де V - кількість газу, що дифундує за одиницю часу; k - коефіцієнт дифузії; S -загальна площа, через яку відбувається дифузія; 1 - товщина мембрани; Р, і Р2 — парціальний тиск газів по обидві сторони мембрани. З урахуванням цього можна виділити такі причини порушень дифузії газів у легенях: 1) зменшення коефіцієнта дифузії. Величина його залежить як від природи газу, так і від середовища, у якому відбувається дифузія. Практично має значення зменшення коефіцієнта дифузії кисню у зв'язку зі зміною властивостей легеневої тканини. При цьому перехід С02 із крові в альвеоли, як правило, не міняється, оскільки коефіцієнт його дифузії дуже високий (у 20-25 разів вищий, ніж кисню); 2) зменшення площі дифузії. Має місце при зменшенні дихальної поверхні легень; 3) збільшення товщини альвеоло-капілярноїмембрани; 4) зменшення різниці між парціальним тиском газів в альвеолярному повітрі та їх напругою в крові легеневих капілярів. Така ситуація виникає при всіх порушеннях вентиляції легень; 5) зменшення часу контакту крові з альвеолярним повітрям. Дифузія кисню порушується в тому випадку, якщо час контакту стає менше 0,3 с. 29.19. Назвіть причини порушень легеневої пєрфузії. Порушення кровообігу в легенях (легеневої пєрфузії) можуть бути зумовлені такими причинами: а) зменшенням тиску в правому шлуночку (недостатність правого серця, зменшення венозного повернення при крововтраті, шоку, колапсі); 6) збільшенням тиску в лівому передсерді (стеноз отвору двостулкового клапана, лі-вошлуночкова недостатність серця); в) збільшенням опору судин малого кола кровообігу. Останнє може бути обумовлено рефлекторним збільшенням тонусу артеріол легень, збільшенням в'язкості крові, наявністю перешкод для руху крові (тромбоз, емболія). 29.20. Які причини і механізми розвитку гіпертензії малого кола кровообігу? Гіпертензія малого кола кровообігу характеризується збільшенням тиску в легеневій артерії понад 25 мм рт. ст. її розвиток може бути обумовлений такими механізмами: а) тривалий спазм артеріол легень. Найчастіше виникає в результаті зменшення парціального тиску кисню в альвеолярному повітрі, що буває при гіпоксичній гіпоксії (див. розд. 19) і порушеннях вентиляції легень; б) гострий рефлекторний спазм легеневих артеріол. Розвивається при емболії судин легень, стенозі отвору двостулкового клапана. В останньому випадку вмикається рефлекс Китаєва: збільшення тиску в лівому передсерді і легеневих венах викликає збудження барорецепторів і спазм легеневих артеріол, що попереджає збільшення гідростатичного тиску в капілярах легень і розвиток набряку; в) збільшення тиску повітря в бронхах і альвеолах. Викликає здавлення легеневих капілярів і, як наслідок, збільшення судинного опору в малому колі кровообігу. Буває у людей під час важких нападів кашлю. При цьому тиск у легеневій артерії може зростати до 250 мм рт. ст.; г) облітерація легеневих судин (артеріол, капілярів, венул) унаслідок ураження їхніх стінок (наприклад, при емфіземі легень). В експерименті показано, що гіпертензія малого кола кровообігу виникає при вимиканні не менше 2/3 судинного русла. Отже, видалення однієї легені не призводить до розвитку цього синдрому; ґ) збільшення хвилинного об'єму серця більше ніж у 3 рази; д) порушення відтоку крові по легеневих венозних судинах (вади двостулкового клапана серця, недостатність лівого шлуночка, здавлювання легеневих вен); є) збільшення в 'язкості крові (наприклад, при поліцитемії); є) уроджені вади, пов'язані зі скиданням крові зліва направо (незарощення Боталло-вої протоки, дефекти міжшлуночкової перегородки). Залежно від того, на якій ділянці легеневих судин збільшується опір, розрізняють пре- і посткапілярну форму гіпертензії малого кола кровообігу.
29.23. Які зміни загальних і місцевих вентиляційно-перфузійних відношень можуть спричинятися до розвитку недостатності зовнішнього дихання? Вешпшяційно-перфузійне відношення для легень у цілому визначається як відношення хвилинного об'єму альвеолярної вентиляції (Va) до хвилинного об'єму крові (Q). У нормі:
Якщо зазначене відношення більше одиниці, то це свідчить про збільшення функціонального мертвого простору, унаслідок чого ефективність вентиляції погіршується. Така ситуація виникає при гіпервентиляції, не підкріпленій збільшенням перфузії, або при нормальній вентиляції, але порушеному легеневому кровообігу. Вентиляційно-перфузійне відношення менше 0,8 свідчить про так званий ефект шунтування, коли кров, не збагачена киснем, потрапляє в легеневі вени і потім у велике коло кровообігу. Це буває, коли величина легеневої перфузії значно перевищує величину альвеолярної вентиляції. В умовах патології у зв'язку з нерівномірністю вентиляції і перфузії різних альвеол вентиляційно-перфузійні відношення в різних ділянках легень можуть бути різними. При цьому вентиляційно-перфузійне відношення для легень у цілому може бути в нормі, хоча і розвиваються ознаки дихальної недостатності. Вони обумовлені існуванням в уражених легенях трьох типів альвеол (рис. 141):
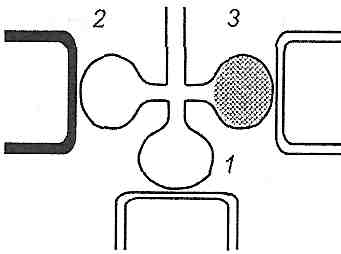
Рис. 141. Місцеві вентиляційно-перфузійні відношення: 1 — ефективний альвеолярний об 'єм; 2 — альвеолярний мертвий простір; 3 — альвеолярний артеріо-венозний шунт 1) альвеоли, які оптимально вентилюються і перфузуються (Va/Q = 0,8... 1). Вони утворюють ефективний альвеолярний об 'єм і становлять більшість альвеол здорових легень; 2) альвеоли, які вентилюються, але не перфузуються (V7Q >1). їхня сукупність становить альвеолярний мертвий простір^ 3) альвеоли, які перфузуються, але не вентилюються (V/Q <0,8). З ними пов'язане артеріовенозне шунтування. Збільшення кількості альвеол другого і третього типів може призводити до розвитку гіпоксемії. При цьому виділення С02 не порушується завдяки високому коефіцієнту його дифузії (розвивається паренхіматозна недостатність дихання).
129. Асфіксія: визначення поняття, причини, патогенез. Термінальне дихання.
Асфіксія (ядуха) - це загрозливий для життя стан, при якому гостра дихальна недостатність досягає такого ступеня, що у кров зовсім не надходить кисень, а з крові не виводиться вуглекислий газ. Найчастіше асфіксія настає у разі здавлення дихальних шляхів (задушення), закупорки їхнього просвіту (сторонні предмети, запальний набряк), наявності рідини в дихальних шляхах і альвеолах (утоплення, набряк легень, потрапляння блювотних мас), у разі двостороннього пневмотораксу. Крім того, асфіксія може розвитися при сильному пригніченні дихального центру, порушенні проведення нервових імпульсів до дихальних м'язів, різкому обмеженні рухомості грудної клітки. У перебігу асфіксії виділяють три періоди. Перший період асфіксії характеризується швидким збільшенням глибини і частоти дихання з переважанням фази вдиху над фазою видиху. Розвивається загальне збудження, підвищується тонус симпатичної частини вегетативної нервової систе- ми (розширюються зіниці, з'являється тахікардія, підвищується артеріальний тиск), можливі судоми. У другому періоді частота дихання поступово зменшується при збереженій максимальній амплітуді дихальних рухів, посилюється фаза видиху. Переважає тонус парасимпатичної частини вегетативної нервової системи (зіниці звужуються, артеріальний тиск знижується, відзначається брадикардія). В третьому періоді асфіксії спостерігають зменшення амплітуди дихання, його частоти і, нарешті, зупинку дихання. Артеріальний тиск значно знижується. Після короткочасної зупинки дихання, як правило, з'являється кілька рідких конвульсивних дихальних рухів (гаспінг-дихання), після яких настає параліч дихання. Явища, що їх спостерігають при асфіксії, пов'язані спочатку з накопиченням в організмі вуглекислого газу. Діючи рефлекторно і безпосередньо на дихальний центр, С02 збуджує його, доводячи глибину і частоту дихання до максимально можливих величин. Крім того, дихання рефлекторно стимулюється і зниженням у крові напруги кисню. У міру збільшення в крові вмісту С02 підвищується і артеріальний тиск. Експерименти з вдиханням газових сумішей, що містять 10-20 % С02, показали, що таке підвищення пов'язане, по-перше, з рефлекторним впливом хеморецепторів на судиноруховий центр, по-друге, з посиленим викидом адреналіну в кров, по-третє, зі збільшенням хвилинного об'єму крові в результаті підвищення тонусу вен і збільшення надходження крові до серця при посиленні дихання. При подальшому збільшенні концентрації С02 у крові починає виявлятися його наркотична дія, рН крові знижується до 6,8-6,5. Посилюється гіпоксемія і, відповідно, гіпоксія головного мозку. Це призводить, у свою чергу, до пригнічення дихання, зниження артеріального тиску. В результаті настає параліч дихання і зупинка серця.
130. Причини і механізми порушення травлення в порожнині рота. Етіологія, патогенез, експериментальні моделі карієсу та пародонтозу. Причини, механізми порушень слиновиділення. Недостатність травлення - це патологічний стан, при якому травна система не забезпечує засвоєння поживних речовин, що надходять в організм. Наслідком цього є розвиток різного ступеня голодування (див. розд. 18). 30.2. Як класифікують недостатність травлення? I. За клінічним перебігом виділяють гостру і хронічну недостатність травлення. II. Відповідно до анатомічного принципу недостатність травлення може бути обумовлена порушеннями цього процесу: а) у порожнині рота; б) у шлунку; в) у кишках. III. Недостатність травлення може бути загальною (тотальною) і селективною (парціальною). При загальній недостатності порушено засвоєння всіх поживних речовин, при селективній - тільки окремих їхніх класів (наприклад, ліпідів, лактози, вітаміну В12 та ін.). IV. За етіологією розрізняють спадково обумовлену (деякі види мальабсорбції) і набуту недостатність травлення. Остання може бути: а) інфекційного походження; б) обумовленою впливами фізичних факторів (наприклад, при гострій променевій хворобі); в) пов'язаною із впливами хімічних агентів; г) диерегуляторною; ґ) аліментарною. V. Патофізіологічний принцип передбачає поділ недостатності травлення на три варіанти. Цс недостатність, обумовлена порушеннями: а) рухової функції травної системи; б) секреторної її функції; в) процесів усмоктування. 30.21. Що може спричинятися до порушень жування? Яке значення для травлення мають ці порушення? Причини порушень жування: 1) ураження зубів та їх відсутність (карієс, пародонтит); 2) ураження жувальних м'язів (міозит); 3) порушення іннервації жувальних м'язів (бульбарні паралічі, неврити); 4) ураження скронево-нижньощелепних суглобів; 5) травматичне ушкодження кісток (нижньої, верхньої щелепи); 6) ураження слизової оболонки порожнини рота і ясен (стоматит, гінгівіт); 7) ураження м'язів і слизової язика; 8) гіпосалівація. Порушення жування мають такі наслідки: Дата добавления: 2016-03-26 | Просмотры: 1084 | Нарушение авторских прав |