|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
ГомоцистинурияМинимальные диагностические признаки: марфаноидный фенотип, повышение концентрации метионина и гомоцистина в плазме крови в сочетании со снижением того же показателя для цистина, а также повышением экскреции гомоцистина с мочой (гомоцистинурией) (рис. 4). В основе патогенеза гомоцистинурии лежит нарушение обмена серосодержащих аминокислот. В норме одна из таких аминокислот — метионин — через ряд промежуточных стадий (в том числе гомо-цистеин и цистатионин) переходит в цистеин. Повышенная концентрация этих естественных для организма веществ вызывает очаговые некрозы в почках, селезенке, слизистой оболочке желудка, сосудах. Повреждения стенок сосудов, а также активизация свертывающей системы крови усиливают тромбообразование, что проявляется тромбозами коронарных, сонных, почечных артерий, генерализованным венозным тромбозом. Это может приводить к артериальной гипертензии, различным неврологическим нарушениям, ранней смерти.
Рис. 4 Гомоцистинурия Галактоземия – отсутствие или существенное снижение активности фермента галактозо-1-фосфат-уридилтрансферазы и накопление в крови галактозы и ее производных, которые оказывают токсическое действие на центральную нервную систему, печень и хрусталик глаза. В первые дни и недели жизни наблюдаются желтуха, увеличение печени, нистагм, гипотония мышц, рвота. Со временем развивается катаракта, отставание в физическом и умственном развитии. Характерна непереносимость молока. Болезнь имеет аутосомно-рецессивный тип наследования. Несколько форм этого заболевания обусловлены различными мутантными аллелями гена галактозо-1- фосфатуридилтрансферазы (рис. 5).
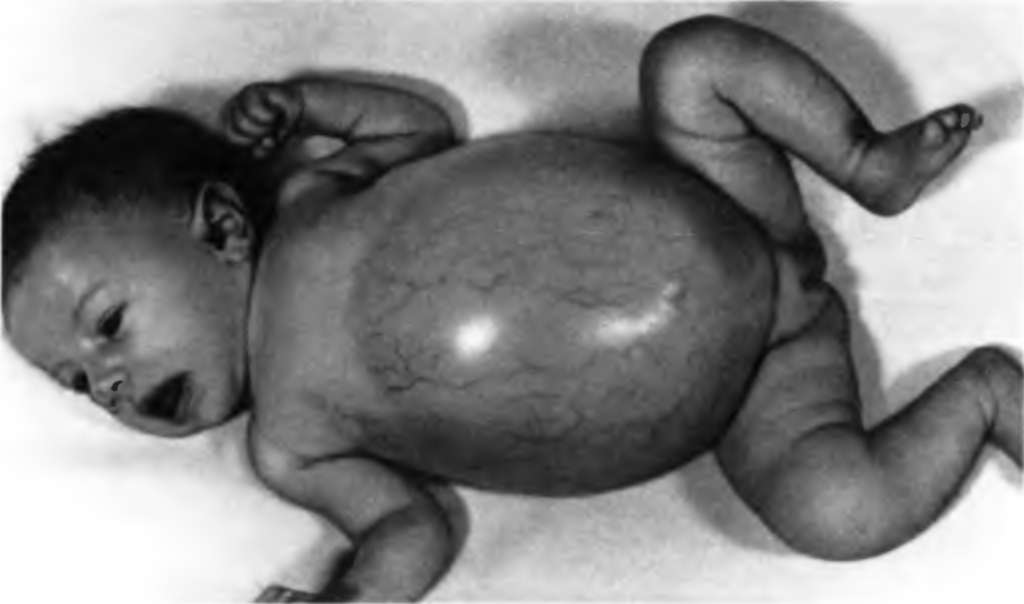
Рис. 5. Галактоземия Болезнь Гирке (гликогеноз I типа, гликогеновая болезнь I типа) – неспособность превращения глюкозо-6-фосфата в глюкозу, которая приводит к нарушению как синтеза, так и разложения гликогена. Депонирование гликогена происходит, обратный процесс – нет. Развивается гипогликемия. Накопление избыточного количества гликогена в печени и почках приводит к печеночной и почечной недостаточности. Тип наследования – аутосомно-рецессивный. Причина заболевания – мутация в гене, который кодирует фермент глюкозо-6-фосфатазу. Описано 14 мутантных аллелей этого гена (рис. 6).
Рис. 6 Болезнь Гирке Дата добавления: 2015-03-04 | Просмотры: 958 | Нарушение авторских прав |