|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
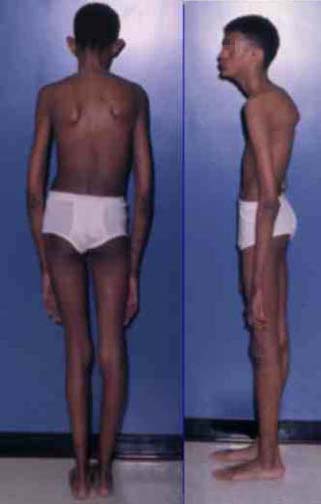
Нарушения обмена соединительной тканиСиндром Марфана («паучьи пальцы», арахнодактилия) – поражение соединительной ткани вследствие мутации в гене FBN1, ответственном за синтез фибриллина. Наследуется по аутосомно-доминантному типу. Клиническая полиморфность заболевания объясняется большим числом мутантных аллелей, каждый из которых может проявляться в гетерозиготном состоянии. Для больных характерен высокий рост, астеническое телосложение (непропорционально длинные конечности), арахнодактилия (длинные тонкие пальцы), слабость связочного аппарата, отслойка сетчатки глаза, подвывих хрусталика, пролапс митрального клапана (рис.10).
Рис. 10 Синдром Марфана Мукополисахаридозы – группа заболеваний соединительной ткани, связанных с нарушеним обмена кислых гликозаминогликанов (мукополисахаридов), вызванных недостаточностью некоторых лизосомных ферментов. Эти заболевания относят к лизосомным болезням накопления. Они проявляются в различных дефектах костной и соединительной тканей. Мукополисазаридоз типа I (синдром Хурлера) – аутосомно-рецессивное заболевание, возникающее в результате дефицита фермента альфа-L-идуронидазы из-за мутаций в гене IDUA. Это приводит к накоплению белково-углеводных комплексов и жиров в клетках организма. В результате у больных наблюдается малый рост, существенная задержка умственного развития, увеличение печени и селезенки, пороки сердца, помутнение роговицы, деформация костей и огрубение черт лица. Мукополисахаридоз типа II (синдром Хантера) – сцепленное с полом рецессивное заболевание, которое обусловлено дефектом фермента идуронатсульфотазы из-за мутации в гене IDS. Веществами накопления являются дерматан- и гепарансульфаты. Характерны грубые черты лица, скафоцефалия, шумное дыхание, низкий грубый голос, частые острые респираторные вирусные инфекции (рис.11). Наблюдаются прогрессирующая тугоухость, узелковые поражения кожи спины, остеоартриты, поражения роговицы.
\ Рис. 11 Синдром Хантера Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо) – заболевание, вызванное накоплением гепарансульфата. Для него характерна генетическая гетерогенность – существуют четыре типа этой болезни, вызванные мутациями в четырех разных генах, кодирующих ферменты, участвующие в метаболизме накапливаемого вещества.. Для больных арактерны задержка роста, контрактуры суставов, гипертрихоз, умеренная гепатоспленомегалия. В отличие от синдромов Хурлера и Хантера при болезни Санфилиппо преобладает умственная отсталость (рис.12).
Рис. 12 Мукополисахаридоз Дата добавления: 2015-03-04 | Просмотры: 707 | Нарушение авторских прав |