|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
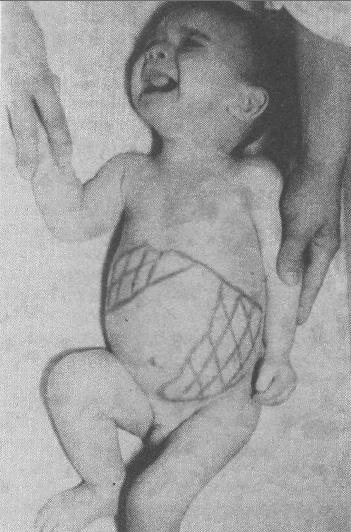
Нарушения липидного обменаБолезнь Ниманна-Пика типов А и Б – снижение активности фермента кислой лизосомальной сфингомиелиназы, который кодируется геном SMPD1. Тип наследования – аутосомно-рецессивный. Нарушение липидного метаболизма приводит к накоплению липидов в печени, легких, селезенке, нервных тканях. Характерна дегенерация нервных клеток, нарушение деятельности нервной системы, повышенный уровень холестерина и липидов в крови. Тип А летален в раннем детском возрасте. Тип Б протекает более мягко, больные, как правило, доживают до взрослого состояния. (рис. 7).
Рис. 7 Болезнь Ниманна-Пика Болезнь Гоше (гликозилцерамидный липидоз) – накопление глюкоцереброзидов в клетках нервной и ретикулоэндотелиальной системы, обусловленное дефицитом фермента глюкоцереброзидазы, которая кодируется геном GBA. Относится к группе лизосомных болезней накопления. Некоторые формы заболевания проявляются в тяжелых поражениях печени, селезенки, нервной и костной тканей (рис. 8).
Рис. 8 Болезнь Гоше Дата добавления: 2015-03-04 | Просмотры: 811 | Нарушение авторских прав |