|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Тема 7.2 Генные болезниГенные болезни встречаются чаще, чем хромосомные. Диагностика этих заболеваний обычно начинается с анализа клинических и биохимических данных, родословной пробанда, типа наследования. Моногенные болезни могут иметь аутосомно-доминантныи, аутосомно-рецессивный и Х-сцеплен-ный типы наследования. При а у т о с о м н о-д о м и н а н т н о м типе наследования патологический признак встречается в каждом поколении родословной и распределение между больными и здоровыми часто составляет 50:50, но вероятность может быть 100, 75 %. Однако пенетрантность патологических проявлений почти всегда ниже 100 %. Клинические проявления заболеваний и их выраженность могут быть различными не только между семьями, но и внутри одной семьи. Кроме того, клинические признаки могут появиться не сразу после рождения, а спустя много лет. При а у т о с о м н о-р е ц е с с и в н о м типе наследования правильному анализу способствуют указания на родственный брак (двоюродный брат и сестра), данные биохимических исследований дефектов обмена веществ (выявление энзимопатий). Научной группой ВОЗ разработана и рекомендована к практическому применению следующая классификация наследственных заболеваний обмена веществ: 1) наследственные нарушения обмена аминокислот (фенилкетонурия и др-); 2) наследственные нарушения обмена углеводов (гликогеновая болезнь, галактоземия и др.); 3) наследственные нарушения обмена липидов (болезни Ниманна - Пика, болезнь Гоше и др.); 4) наследственные нарушения обмена стероидов (адреногенитальный синдром и др.); 5) наследственные нарушения обмена пуринов и пиримидинов (синдром Леша—Найяна и др.); 6) наследственные нарушения обмена соединительной ткани (мукополи-сахаридозы, синдром Марфана и др.); 7) наследственные нарушения обмена в эритроцитах (анемия Минков-ского — Шоффара и др.); 8) наследственные нарушения всасывания в пищеварительном тракте (муковисцидоз, целиакия, непереносимость лактозы и др.). Рассмотрим наиболее часто встречающиеся ферментопатии, возникающие в результате генных мутаций. ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С РАССТРОЙСТВОМ АМИНОКИСЛОТНОГО ОБМЕНА Фенилкетонурия (ФКУ, фенилпировиноградная олигофрения, болезнь Феллинга). Это заболевание обусловлено биохимическим дефектом превращения аминокислоты фенилаланина. Тип наследования аутосомно-рецессив-ный. Больные являются гомозиготами по гену фенилкетонурии, а родители гетерозиготами. Биохимический дефект состоит в ферментном (энзимном) блоке нормального превращения фенидаданина_в аминокислоту теитозин из-за недостаточности фермента фенилаланингидроксилазы. Количество фенилаланина в организме накапливается и концентрация его в крови увеличивается в 10 — 100 раз. Далее он превращается в фенилпировиноградную кислоту, являющуюся нейротропным ядом. Накопление фенилаланинав организме идет постепенно и клиническая картина развивается медленно. В первом полугодии жизни у ребенка бывают срыгивания, могут развиваться дерматиты и судорожные припадки. Судорожный синдром развивается по типу малой эпилепсии. В последующем соматическое развитие ребенка мало страдает, но психическое развитие, моторика все больше отстают или деградируют. Только 0,5 % больных сохраняют нормальный интеллект. В характере выявляется импульсивность, резкая возбудимость, склонность к агрессии. Почти все дети блондины с голубыми глазами С мочой и потом выделяются продукты обмена фенилаланина (фенилуксусная кислота) и от ребенка исходит неприятный запах («мышиный», «волчий», «затхлый»). Частота этого заболевания составляет 1 на 5600 новорожденных. Исключение из питаня фенилаладина с первых месяцев жизни способствует нормальному развитию ребенка. Внастоящее время все новорожденные обследуются на уровень фенилаланина в крови: для этого несколько капель крови на фильтровальной бумаге посылают в лабораторию, где с помощью хроматографического метода определяют содержание данной аминокислоты. Реже используется проба Феллинга: к 2- -5 мл свежей мочи ребенка добавляют 10 капель 10 % раствора треххлористого железа. Появление сине - зеленого окрашивания свидетельствует о наличии заболевания и ребенок должен быть обследован количественными методами для установления окончательного диагноза. ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С НАРУШЕНИЕМ ОБМЕНА УГЛЕВОДОВ Одной из, основных частей пищи человека являются углеводы, которые выполняют энергетическую и структурную функции и являются составными частями некоторых биологически активных веществ (ферментов, гормонов, иммунных тел), мукополисахаридов. Основным углеводом пищи для детей грудного возраста является лактоза, а в последующем — крахмал. Представителем в нарушении межуточного обмена углеводов является гликогеиовая болезнь. Гликогеновая болезнь. Характерной особенностью этого заболевания является нарушение энзимного звена в сложной цепи процессов, связанных с синтезом и разложением гликогена (животного крахмала). В норме образующийся из глюкозы гликоген при голодании должен снова превратиться в глюкозу и только в этом виде организм человека может использовать углеводы для других целей. Различают несколько типов гликогенозов. Первый тип гликогеноза — гепатореналъный, или болезнь Г и р к е. При этом заболевании накопившийся гликоген в печени и почках не может обратно превратиться в глюкозу и в организме развивается гипогликемия, так как в печени отсутствует фермент глюкозо-6-фосфатаза, которая играет важную роль в регуляции уровня глюкозы в крови. Наследуется заболевание по аутосомно-рецессивному типу. В различные возрастные периоды клиническая картина болезни Гирке неодинакова. В период новорожденности основными симптомами являются гипогликемические судороги и гепатомегалия. Задержка роста начинает отмечаться с 1-го года жизни. Характерен внешний вид больных: большая голова, «кукольное» лицо, короткая шея, выступающий живот. Уровень глюкозы в крови почти всегда ниже нормы, а липидов и молочной кислоты увеличен. На почве гипогликемии отмечаются обморочные состояния или.судороги. Умственное развитие почти не изменяется, подовое развитие задерживается. В крови повышается уровень мочевой кислоты и после пубертатного периода нередко наблюдаются признаки подагры. При нагрузке глюкозой отмечается либо нормальная гликемическая кривая, либо диабетическая, когда уровень сахара в крови к 90—120 мин не приходит к исходному уровню и имеется высокий подъем. При введении адреналина уровень глюкозы в крови увеличивается незначительно. Диагноз подтверждают после исследования биопсийного материала печени. В лечебном плане большое значение уделяют диетотерапии. Частоту приемов пищи необходимо увеличить для предупреждения выраженной гипогликемии. В этих же целях повышают количество углеводов в диете. Жир ограничивают, белки назначают в соответствии с возрастной нормой. Лечение глюкагоном несколько улучшает прогноз заболевания. Второй тип гликогеноза, или болезнь Помпе, самый неблагоприятный. Его называют генерализованным, так как накопление гликогена происходит не только в печени, но и в скелетных мышцах, миокарде, легких, селезенке, надпочечниках, стенках сосудов, нейронах. У новорожденных детей размеры сердца нормальные. Спустя 1—2 месяца у ребенка появляется мышечная слабость, теряются уже имеющиеся двигательные навыки. При кормлении и плаче отмечается цианоз. В это время выявляются кардиомегалия и макроглоссия. Сухожильные рефлексы исчезают, но плотность мышц остается нормальной. Накопившийся секрет в дыхательных путях приводит к затяжному течению пневмонии, а затем и к гибели больного. Диагностика заболевания возможна еще до рождения ребенка. Для этого определяют активность специфических ферментов в амниотической жидкости и ее клетках (определение кислой α-1,4-глюкозидазы). Третий, четвертый, пятый, шестой и другие типы гликогеноза напоминают клиническую картину первого типа и без биохимических исследований дифференциальная диагностика затруднительна. Галактоземия. Это заболевание характеризуется накоплением в крови галактозы и проявляется отставанием в физическом и умственном развитии, тяжелым поражением печени, нервной системы, глаз и других органов. Наследуется галактоземия по аутосомно-рецессивному типу, частота патологии составляет 1 из 16000. Галактоза является составной частью молочного сахара лактозы, при гидрелизе которой в пищеварительном тракте образуются глюкоза и галактоза. Галактоза тормозит всасывание глюкозы и этим создает углеводную среду в кишечнике. Она необходима для миелинизации нервных волокон. Однако избыточные ее количества для организма нецелесообразны, и поэтому она превращается в глюкозу с помощью фермента галактоза-1-фосфат-уридил-трансферазьг. При низкой активности этого фермента происходит накопление галактоза-1-фосфата, который оказывает токсическое действие на функцию печени, мозга, хрусталик глаза. Начало заболевания может проявляться с первых дней жизни расстройствами пищеварения, интоксикацией (понос, рвота, обезвоживание), развитием гипотрофии. Печень увеличивается, при пальпации она плотная, появляется желтуха, нарастают признаки печеночной недостаточности. Обнаруживается помутнение хрусталика глаза (катаракта). При тяжелом течении и без лечения дети погибают на первом году жизни, а при вскрытии обнаруживают цирроз печени. У выживших отмечается резкое отставание психомоторного развития, гепатомегалия, катаракта. Наиболее точным методом диагностики галактоземии является исследование в эритроцитах ферментов галактоза-1-фосфата и галактоза-1-фосфатуридилтрансферазы, галактозы в крови и моче, где уровни ее увеличены. Исключение из пищи молока (источника галактозы) дает возможность нормально развиваться больному ребенку. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С НАРУШЕНИЕМ ЛИПИДНОГО ОБМЕНА В группу липидов входят триглицериды, холестерол (холестерин), эфиры холестерола, фосфолипиды, сфинголипиды, свободные или неэстерифицированные жирные кислоты (НЭЖК) и др. Нарушение обмена липидов может происходить на уровне расщепления, всасывания, транспорта, а также при межуточном обмене. Исследования показали, что можно выделить два основных типа наследственных нарушений обмена липидов: 1) липидозы или сфинголипидозы — болезни, приводящие к накоплению сфинголипидов в клетках разных тканей (внутриклеточные липидозы); 2) болезни с нарушением липопротеидов, содержащихся в крови. Среди наследственных заболеваний, приводящих к накоплению липидов внутри клеток, наиболее изучены болезнь Ниманна - Пика, Гоше и амавро-тическая идиотия. Болезнь Ниманна—Пика. Это заболевание связано с накоплением сфингомиелина в мозге, печени, ретикуло-эндотелиальнои системе. Сущность нарушения обмена сфингомиелинов состоит в утрате ферментативной активности сфингомиелиназы. Тип наследования аутосомно-рецессивный. В патогенетическом плане заболевание связано с включением в состав сфитчмиелина жирных кислот, что не свойственно нормальному сфингомиелину. Атипичный сфингомиелин накапливается в клетках. Заболевание развиваетсяв в раннем детском возрасте с отказа от еды, появления рвоты, увеличения живота, печени, селезенки
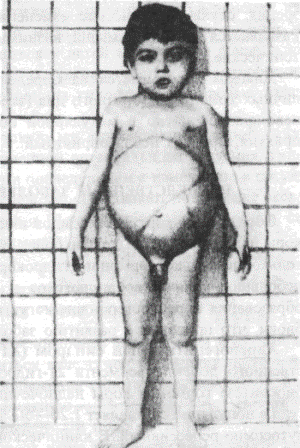
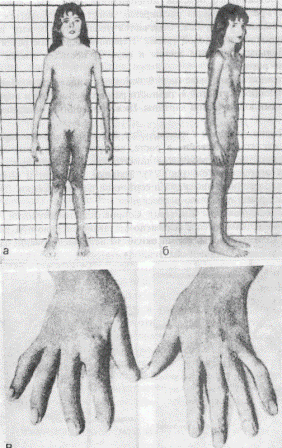
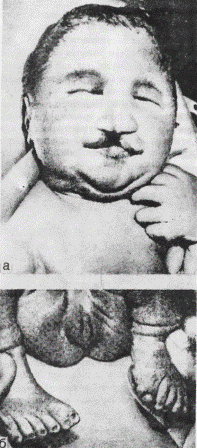
У таких больных отмечаются мышечная слабость, судороги, снижается слух и зрение. На глазном дне у 20—30 % детей отмечается пятно вишневого цвета. В анализах крови — анемия, тромбоцитопения, вакуолизированные лимфоциты, увеличение холестерина, лецитина, сфингомиелина. Прогноз заболевания неблагоприятный. Дети погибают в раннем возрасте. Болезнь Гоше. При этом заболевании в клетках нервной, рети-, кулоэндотелиальной системы накапливаются цереброзиды. В основе болезни Гоше лежит утрата активности фермента глюкоцеребро-зидазы, приводящая к накоплению в клетках ретикуло-эндотелиаль-ной системы глюкоцереброзида. Различают детскую и ювенильную формы. В пунктате костного мозга, селезенки находят крупные клетки Гоше. Накопление цереброзидов в клетках нервной системы приводит к их разрушению. Детский тип проявляется в первые месяцы жизни задержкой умственного и физического развития, увеличением живота, печени и селезенки, развитием дыхательной недостаточности (инфильтрация легких клетками Гоше), гипертонией мышц, наблюдаются судороги и ребенок погибает на первом году жизни. Ювенильная форма поражает детей различных возрастов и заболевание носит хронический характер. В клинической картине отмечаются анемия, спленомегалия, пигментация кожи (коричневые пятна), остеопороз, переломы и деформация костей. Развитие иммунодефицитных состояний приводит к смерти. Амавротическая идиотия (болезнь Тей—Сакса). Это заболевание связано с резким увеличением в клетках мозга, а также печени и селезенки ганглиозидов из-за дефицита гексозаминидазы А в организме. При рождении и в первые 3—4 месяца жизни дети не отличаются от здоровых сверстников. Заболевание развивается медленно, ребенок становится менее активным, теряет приобретенные навыки. Рано появляются расстройства зрения, слуха. Психические изменения прогрессируют вплоть до идиотии. Развивается гипотония мышц, возникает паралич конечностей. Часто бывают тонические судороги. Диагноз основывается на определении активности гексозаминидаз, типичных изменениях глазного дна (атрофия сосков зрительных нервов, вишнево-красное пятно в макулярной области). Несмотря на лечебные мероприятия, прогноз неблагоприятный. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ ОБМЕНА СТЕРОИДОВ Образование кортикостероидов имеет сложный ферментативный путь. Из коры надпочечников выделено более 40 соединений, однако в кровь поступает только несколько гормонов: гидрокортизон, альдостерон, кортикостерон и небольшое количество андрогенов и эстрогенов. На промежуточном пути образования кортикостероидов могут возникать различные ферментативные блоки, что приводит к развитию заболеваний. Адреногенный синдром (АГС). Развивается в результате наследственного дефекта фермента 21-гидроксилазы, приводящего к нарушению биосинтеза гормонов коры надпочечников. Распространенность мутантного гена в популяции составляет 1:20—50. Тип наследования данного заболевания аутосомно-рецессивный. В клинической картине выделяют три формы: соль-теряющую, вирильную и форму с гипертензией. Солътеряющая форма развивается в первые недели жизни. Проявляется отказом ребенка от груди, рвотой, а иногда и учащением стула, что способствует значительному обезвоживанию организма. При этом у ребенка могут развиться коллаптоидное состояние, судороги, нарушение сердечной деятельности, что приводит к летальному исходу. Лабораторное исследование сыворотки крови выявляет гипонатриемию. Типохлоремию и гиперкалиемию. Дифференциально-диагностическое значение имеет гиперкалиемия, так как при пилоростенозе, кишечном токсикозе с эксикозом всегда отмечается низкий уровень калия в сыворотке крови. У новорожденных девочек отмечается различная степень маскулинизации от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с формированием мошонки. Внутренние половые органы таких больных сформированы правильно, кариотип – 46XX. Отмечается гиперпигментация окодатсковой и генитальной областей. У мальчиков основным клиническим симптомом является преждевременное половое развитие, закрытие зон роста эпифизов, в связи с чем больные имеют низкий рост. Во всех случаях отмечается повышение в моче уровня 17-кетостероидов. Если таким больным не оказывается правильная и своевременная терапевтическая помощь (введение гидрокортизона, физиологического раствора), то заболевание заканчивается смертью. На вскрытии, помимо общего истощения, обезвоживания, специфичным является обнаружение увеличенных надпочечников. Вирильная форма АГС развивается вследствие избыточного образования андрогенов при недостатке гидрокортизона. Значительное увеличение клитора, полового члена, оволосение по мужскому типу характеризуют эту форму АГС, которая может наблюдаться у детей младшего и дошкольного возраста. Истинное половое развитие задерживается, так как андрогенные гормоны тормозят секрецию гонадотропина гипофизом и половые железы не развиваются. Гипертоническая форма имеет такую же клиническую картину, как и вирильная, но сопровождается увеличением артериального давления, гипертрофией левого желудочка, что ведет в последующем к изменению в сосудах, поражению почек. Лабораторные исследования выявляют повышение экскреции с мочой 17-оксикортикостероидов и 17-гидроксикортикостероидов, снижение в крови кортизола и альдостерона. НАСЛЕДСТВЕННЫЕ НАРУШЕНИЯ БИОСИНТЕЗА ТИРЕОИДНЫХ ГОРМОНОВ Гипотиреоз. Это заболевание, обусловленное понижением функции щитовидной железы. Оно может развиться вследствие поражения щитовидной железы — первичный гипотиреоз, поражения гипоталамо-гипофизарной области, регулирующей тиреоидную функцию, — вторичный гипотиреоз. Гипотиреоз может быть как врожденный, так и приобретенный. Первичный врожденный гипотиреоз чаще возникает в результате внутриутробного порока развития щитовидной железы (аплазия, гипоплазия и др.) или из-за генетического дефекта в синтезе тиреоидных гормонов. Больные дети рождаются с большой массой тела, что связано с отеком тканей. После рождения у ребенка длительно держится желтуха- Они плохо растут и имеют малую прибавку массы тела, отстают в нервно-психическом развитии, отмечаются запоры. Кожа при гипотиреозе сухая, бледная, шелушащаяся, холодная на ощупь. Частота сердечных сокращений ниже нормы, границы сердца расширены. У многих больных имеется систолический шум. Отмечаются грубый голос и скудный рост волос на голове. Рентгенологическое исследование костей запястья выявляет отставание костного возраста. Для диагностики используют исследование в крови гормонов щитовидной железы (тироксина и трийодтиронина), а также тиреотропного гормона гипофиза. Вспомогательное значение имеет повышение уровня холестерина в крови (более 6,8 ммоль/л). Гипотиреоз может быть и у взрослых людей, клиническими проявлениями которого служат заторможенность, гиподинамия, бледность, сухость и отечность кожи, брадикардия, снижение температуры тела, артериального давления. После инфекций, охлаждения, приема снотворных препаратов и проведения оперативных вмешательств может развиться гипотиреоидная (мик-сематозная) кома. Наследование гипотиреоза аутосомно-рецессивное. Лечение гипотиреоза проводят с помощью заместительной терапии препаратами щитовидной железы, начиная с малых доз. Можно использовать и синтетические препараты — тироксин и трийодтиронин. При недостатке тиреотропного гормона используют его синтетический аналог. Гипотирео-идная кома требует неотложных мероприятий, в том числе и введения гидрокортизона. При своевременной диагностике и адекватной терапии прогноз для больных с гипотиреозом благоприятный. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ СОЕДИНИТЕЛЬНОЙ ТКАНИ Соединительная ткань выполняет в организме опорную, трофическую и защитную функции, обеспечивает постоянство внутренней среды живого организма. Выделяют три компонента соединительной- ткани: а) клеточные элементы, б) коллагеновые, эластические и ретикулярные волокна, в) аморфное основное вещество. Сложная структура соединительной ткани имеет генетическую природу и дефекты ее биосинтеза и распада приводят к развитию различных наследственных заболеваний. Болезнь Марфана. Это наследственное заболевание, характеризующееся 2-системным поражением соединительной ткани. Тип наследования — ауто-сомно-доминантный. В развитии этого заболевания имеет значение поражение1 эластина и коллагена, выражающееся в нарушении внутри- и межмолекулярных связей в этих структурах. Для больных типичны высокий рост, длинные (паукообразные) пальцы, воронкообразная или килевидная грудная клетка, плоскостопие (рис. 24). Нередко встречаются бедренные и паховые грыжи, гипоплазия мышц и подкожной клетчатки, мышечная гипотония. При обследовании выявляются врожденные пороки сердца, а с возрастом развивается расслаивающаяся аневризма аорты. Зрение у таких больных снижено, при осмотре выявляется миопия, отслойка сетчатки, подвывих хрусталика, катаракта, косоглазие. В моче определяется повышенное количество мукополисахаридов и их составных частей, которые играют важную роль в формировании коллагена и эластических волокон. Лечение проводят только симптоматическое. Значительные деформации грудной клетки требуют оперативного лечения, что способствует лучшему функционированию сердца, а также улучшению общего самочувствия, показателей электрокардиограммы. Мукополисахаридозы. Гаргоилизм. Для заболеваний этой группы общим является нарушение метаболизма кислых гликозаминогликанов. Это приводит к тому, что патологические продукты обмена откладываются в соединительной ткани, а также в печени, селезенке, роговице, клетках центральной нервной системы. Название «гаргоилизм» произошло от французского слова «gargoille», которым называли уродливые фигуры, украшающие собор Нотр-Дам в Париже
В клинической картине заболевания уже в первые месяцы жизни отмечаются типичные изменения конфигурации черепа («башенный» череп), грубые черты лица с крупными губами и языком, короткая шея, запавшая переносица, укороченное туловище. В дальнейшем развивается кифоз грудного или поясничного отделов позвоночника (рис. 25). Эпифизы костей утолщены. Границы сердца расширены, могут выслушиваться шумы. Окружность живота увеличена, отмечается увеличение печени и селезенки, наличие пупочной и паховой грыжи. Нервно-психическое развитие отстает. У таких больных снижены зрение и слух. В процессе жизни развивается помутнение роговицы. В настоящее время выделяют 8 основных типов мукополисахаридозов, характеризующихся утратой активности разных ферментов и определенными особенностями клинической картины. Ниже приводится описание некоторых из них. Первый тип (синдром Гурлера). Для данного синдрома характерны деформации скелета, отставание в росте, контрактуры крупных суставов, гепатоспленомегалия, кардиомегалия, помутнение роговицы, прогрессирующая умственная отсталость Больные погибают в возрасте до 10 лет от бронхолегочных инфекций и сердечной декомпенсации. Наследование данного синдрома аутосомно-рецессивное. Второй тип (синдром Хантера). Сопровождается глухотой, костно-суставными изменениями, менее выражено отставание нервно-психического развития. Продолжительность жизни около 30 лет. Наследование синдрома —
рецессивное, сцепленное с Х-хромосомой, поэтому болеют только мальчики. Третий тип (синдром Сапфшиппо). Характеризуется умственной отсталостью, незначительными гепатоспленомегалией и катарактой, менее женными костными изменениями. Естественно, расшифровка типа мукополисахаридоза возможна только при помощи биохимических исследований кислых гликозаминогликанов в крови и моче. НАСЛЕДСТВЕННЫЕ НАРУШЕНИЯ ОБМЕНА В ЭРИТРОЦИТАХ Наследственный микросфероцитоз (семейная гемолитическая анемия Минковского—Шоффара). Заболевание обусловлено генетическим дефектом эритроцитов, в частности врожденной недостаточностью липидов оболочки, что приводит к проникновению в клетку ионов натрия и потере АТФ. Измененные эритроциты разрушаются в селезенке, в результате чего происходит образование токсического непрямого билирубина. Заболевание характеризуется триадода синдромов: анемия, желтуха и спленомигалия. В клинической картине следует выделять хроническое течение с триадой синдромов и острые формы, связанные с усиленным гемолизом., Особые трудности для диагностики представляет болезнь в периоде новорожденности. Изучение родословной в таких случаях оказывает существенную помощь. Для диагностики используют исследование крови: в анализах отмечаются микросфероцитоз, ретикулоцитоз, снижается осмотическая стойкость эритроцитов, изменяется структура кислотной эритрограммы. Тип наследования — аутосомно-доминантный. При наличии заболевания у одного из родителей риск рождения ребенка с гемолитической анемией Минковского—Шоффара составляет 50 %. В лечебном плане проводят симптоматические мероприятия, а при гемолитических кризах радикальным методом лечения является спленэктомия. НАСЛЕДСТВЕННЫЕ СИНДРОМЫ НАРУШЕННОГО ВСАСЫВАНИЯ К данной группе энзимопатий относятся непереносимость лактозы, целиакия, муковисцидоз и др. Непереносимость лактозы. Это наследствётгное заболевание, связанное с отсутствием или снижением активности фермента лактазы, расщепляющей молочный сахар до глюкозы и галактозы. Негидролизованная лактоза почти не всасывается и ускоряет перистальтику кишечника. При лактазной недостаточности тонкой кишки с первых дней жизни у ребенка наблюдается упорное послабление стула, беспокойство (колики), срыгивание, обезвоживание, отсутствие прибавки в массе тела. В крови отмечаются значительные электролитные нарушения (снижение уровней калия, натрия, кальция). Нагрузка лактозой выявляет низкий (менее 1,1 ммоль/л) подъем уровня гликемии (при сравнении с исходной концентрацией), рН кала сдвигается в кислую сторону. Непереносимость лактозы встречается у 15 % здоровых детей, а при различных хронических заболеваниях пищеварительного тракта у 40—78 %. Исследования на близнецах и их родителях показали, что наследуется данный ферментный дефект по аутосомно-доминантному типу. Лечебные мероприятия связаны с симптоматической терапией (ликвидация обезвоживания), а также переводом ребенка на безмолочное вскармливание (детские смеси, приготовленные на основе сои). Целиакия (болезнь Ги—Гертера—Гейбнера, глютеновая энтеропатия). Это наследственное заболевание, обусловленное дефицитом ферментов, расщепляющих глиадин злаков до аминокислот, и накоплением в организме токсигенных продуктов его неполного гидролиза. Ведущим клиническим признаком данного заболевания является тяжелая дистрофия, а также увеличение живота, учащение стула, анорексия, отставание в росте. Первые признаки заболевания появляются после включения в питание ребенка блюд, приготовленных из круп, муки злаковых (рожь, пшеница, овес, ячмень). Наследование заболевания — аутосомно-рецессивное. Частота встречаемости этой патологии составляет от 1: 800 до 1: 3000. В родословной семьи, имеющей детей с целиакией, можно выявить хронические заболевания пищеварительного тракта, бесплодие, онкологические заболевания пищеварительного тракта и женских половых органов. В лечебном плане необходимо исключить из питания блюда из злаковых продуктов и проводить симптоматическую терапию. Так как при целиакии всегда имеется дефицит цинка, то его назначение дает положительный эффект. Аглиадиновую диету необходимо практически соблюдать всю жизнь. Муковисцидоз (кистофиброз поджелудочной железы). Это наследственное заболевание, причиной которого является нарушение секреторной функции всех эндокринных желез, выражающееся в повышении вязкости секрета. Тип наследования муковисцидоза — аутосмно-рецессивный. Частота заболеваемости по данным различных авторов от 1из 2000 до 1 из 2500. Выделяют следующие основные клинические формы заболевания: 1) мекониальный илеус (у новорожденных); 2) бронхолегочная форма с развитием хронического воспалительного процесса и потерей массы тела; 3) желудочно-кишечная форма с развитием дистрофии; 4) смешанная форма. Среди всех случаев муковисцидоза мекониальный илеус составляет 5—10 %. В просвет кишечника мало поступает трипсина и меконий не подвергается ферментативному воздействию, в результате чего он становится очень вязким, часто скапливается в илеоцекальной области, прилипает к стенкам кишечника. У ребенка после рождения появляется рвота с желчью, вздутие живота, отсутствие выделения первородного кала, может произойти заворот или перфорация кишечника. При лечении этой формы муковисцидоза применяют панкреатические ферменты, назначают очистительные клизмы, а при перфорации или завороте кишечника — оперативное лечение. Бронхолегочная форма муковисцидоза проявляется повторными заболеваниями легких, трудно поддающимися лечению. При этой форме нередко у ребенка развивается дистрофия. Наряду с поражением легких имеется нарушение переваривающей способности поджелудочной железы. В кале обнаруживается нейтральный жир, стул обильный с неприятным запахом. При желудочно-кишечной форме нарушается переваривающая способность ферментов поджелудочной железы и кишечника. Потребление большого количества пищи с достаточным количеством белков, липидов, углеводов и витаминов не ликвидирует дистрофию. У больных муковисцидозом могут быть и непостоянные симптомы, серо-коричневая окраска зубов, неприятный запах изо рта, длительные боли в верхней половине живота, выпадение прямой кишки, отеки, поражение печени, синуситы. Для диагностики заболевания в периоде новорожденности можно исследовать меконий на концентрацию в нем белка — альбумина (в норме содержание альбумина в меконий до 20 мг/г сухой массы). У более старших детей исследуют потовую жидкость на концентрацию натрия и хлора. Потовую жидкость получают с помощью электрофореза с пилокарпином. Для муковисцидоза характерно увеличение в 2—5 раз концентрации электролитов в поту (норма 35 ммоль/л). Лечение симптоматическое. Назначают диету, обогащенную белком и натрием, с ограничением липидов. С заместительной целью назначают ферментные препараты поджелудочной железы (панкреатин и его аналоги), пищу подсаливают. Смешанная форма муковисцидоза сочетает в себе симптомы бронхолегочной и желудочно-кишечной формы. Изложенные наследственные заболевания обмена веществ, естественно, не исчерпывают все встречающиеся нозологические формы. Дальнейшее развитие клинической генетики и биохимии позволит расшифровать патогенез многих известных наследственных заболеваний, а также описать новые их формы, разработать рациональные методы лечения.
Синдром описан Клайнфельтером в 1942 г., а в 1959 г. была установлена его хромосомная природа (47. ХХY). Вбуквальном соскобе обнаруживаются тельца Барра (Х-половой хроматин). Частота встречаемости составляет 2 из 1 000 новорожденных мальчиков. Однако клинически аномалия проявляется после пубертатного периода. Обязательный признак заболевания — гипоплазия гонад. Для мужчин с синдромом Клайнфельтера характерен высокий рост, пропорции тела — евнухоидные. Скелет развит по женскому типу (широкий таз, узкие плечи), отмечается гинекомастия и ожирение, слабый рост волос на лице (или он отсутствует); на лобке отмечается рост волос по женскому типу. Половой член уменьшен или нормальных размеров, нарушен сперматогенез и в результате этого мужчины бесплодны. Умственное развитие отстает, и до наступления периода полового развития этот признак является ведущим. Однако встречаются лица и с нормальным интеллектом. Помимо хромосомного набора 47, XXY могут быть лица с синдромом Клайнфельтера и с набором хромосом 48, ХХХY, 49, ХХХХY и др. Замечено: чем больше число Х-хромосом, тем больше умственная отсталость, вплоть до итиотии. Изменение кариотипа может быть и в виде 47, ХYY, 48, ХХYY. У этих лиц помимо клинических черт синдрома Клайнфельтера в поведении могут наблюдаться агрессивность, немотивированные поступки. Синдром трисомии X. Впервые синдром трисомии по Х-хромосоме был описан в 1959 г., когда в ядрах эпителия слизистой оболочки щеки больной было обнаружено два тельца полового хроматина. Клинически картина этого заболевания чрезвычайно разнообразна. Такие женщины, как правило, имеют недоразвитые яичники, гипоплазию матки, нерегулярный менструальный цикл, бесплодие, у них рано наступает вторичная аменорея. Однако около 30 % таких больных сохраняют генеративную функцию и могут иметь детей. У женщин с трисомиеи X довольно часто имеется незначительное снижение интеллекта. Отмечена повышенная вероятность развития у таких больных психозов: наиболее часто обнаруживается шизофрения с неблагоприятным типом течения. Примерно у одной трети женщин с кариотипом 47, XXX описаны умеренно выраженные соматические аномалии, высокий рост, кифосколиоз, укорочение и искривление мизинцев на руках, черепнолицевые диспропорции. Три тетра- и пентасомии-Х характерны тяжелые нарушения интеллекта, выраженные соматические аномалии, недоразвитие гениталий. Предварительный диагноз синдрома трисомии X основан на исследовании полового хроматина, а окончательный устанавливается только после определения кариотипа. Лечение в основном симптоматическое и в первую очередь направлено на устранение нарушений функции яичников. СИНДРОМЫ С ЧИСЛОВЫМИ АНОМАЛИЯМИ АУТОСОМ Синдром Дауна. Данная аномалия является самой частой формой хромосомной патологии человека и проявляется трисомиеи по^^Г^паде^ хромосом. Заболевание встречается с частотой 1 из 700—800 новорожденных. Простая трисомия составляет около 95 % от общего числа больных с синдромом Дауна, а 4 % приходится на транслокационный вариант и 1 % на мозаицизм. В основе болезни Дауна лежит нерасхождение по 21-й паре хромосом либо в яйцеклетке во время мейоза, либо на ранних стадиях дробления зиготы. Кариотип больного при трисомии содержит 47 хромосом, при этом лишней является 21-я хромосома. При транслокационном варианте в кариотипе содержится 46 хромосом, а лишняя 21-я хромосома оказывается транслоцированной чаще всего на хромосому группы D или G. Иногда подобная транслокация в сбалансированном состоянии обнаруживается у одного из родителей (чаще всего у матери). Для такой семьи имеется повышенный риск повторного рождения ребенка с болезнью Дауна, так как в мейозе у таких родителей наряду с нормальными гаметами будут возникать гаметы с несбалансированным кариотипом. Как правило, клиническая картина трисомного и транслокационного вариантов неразличима. При мозаичном варианте (норма—трисомия) выраженность клинических симптомов болезни Дауна зависит от соотношения нормального и патологического клонов: чем меньше процент нормальных клеток с 46 хромосомами, тем более выражена клиническая картина.
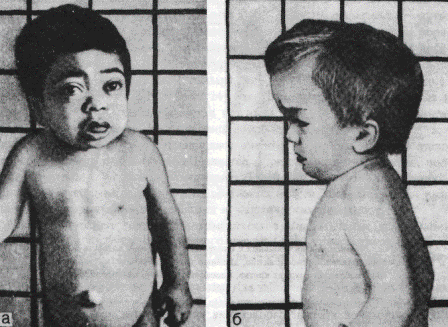
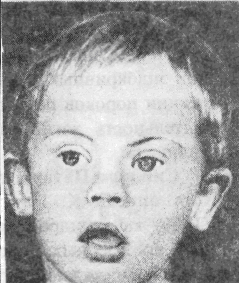
Мальчики и девочки с болезнью Дауна рождаются с одинаковой частотой. Масса и длина тела при рождении обычно соответствуют доношенному ребенку. Голова меньших размеров со скошенным затылком. Лицо плоское, с косым монголоидным разрезом глаз, широкой переносицей маленьким носом, большим языком часто не вмещающимся во рту. У больных рот полуоткрытый, на губах часто трещины, могут быть аномалии зубов, ушных раковин. Суставы имеют большую подвижность, пальцы короткие, на ладони пролегает глубокая борозда («обезьянья борозда»).Мьшцы гипотоничные, живот увеличен часто имеются врожденные пороки сердца. Грудная клетка деформирована. Умственное развитие больных отстаёт, возможно развитие тяжелой идиотии. Синдром Дауна сопровождается расстройствами эндокринных желез и нарушением обмена веществ. Продолжительность жизни больных с синдромом Дауна ограничена. Однако при нормализации эндокринных функций и коррекции пороков развития продолжительность жизни может быть удлинена. Синдром Патау. Этот синдром был описан К. Патау с соавт. (1960) как синдром множественных врожденных пороков развития, сопровождающийся трисоми-ей гю 13-й хромосоме (рис. 21). При рождении эти дети имеют малую массу тела, хотя рождаются в срок, у беременных ими женщин отмечается многоводие. Характерен внешний вид больного: окружность черепа уменьшена, низкий лоб, узкие глазные щели, запавшая переносица, типична расщелина губы, нёба. Характерная микрофтальмия, помутнение роговицы. Из аномалий косшо - мышечной системы наиболее постоянны полидактилия и флексорное положение кистей. Интеллект нарушен, 95 % таких больных умирают в возрасте до года, этому способствуют врожденные пороки сердца (дефекты межпредсердной и межжелудочковой перегородки), органов пищеварения, поликистоз почек. При синдроме Патау всегда поражены гениталии: у мальчиков обычно отмечается крипторхизм, а у девочек дупликация матки и влагалища. Синдром «кошачьего крика». Этот синдром связан с делецией короткого плеча 5-й хромосомы (описан в 1963 г.). Плач новорожденных похож на крик кошки, что связано с аномалиями развития гортани и голосовых связок. Дети плохо растут, отстают в психическом развитии. Внешний вид больных имеет особенности: микроцефалия, лицо круглое с гипертелоризмом, микрогнатия, эпикант, уши неправильные и низко расположенные, короткая шея. Врожденные пороки внутренних органов встречаются сравнительно редко, наиболее часто порочно развитым оказывается сердце. Большинство детей умирает в раннем возрасте, однако описаны больные старших возрастов, в частности – 53 летняя женщина. Таким образом, изложенная клиническая картина заболеваний при различных хромосомных нарушениях сопровождается в первую очередь отставанием в умственном развитии и множеством пороков развития. Предположительная диагностика возможна на основании клинической картины, а окончательный диагноз устанавливается только после исследования хромосомного набора. Всем этим больным необходима консультация врача-генетика. Дата добавления: 2015-12-16 | Просмотры: 2535 | Нарушение авторских прав |