|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
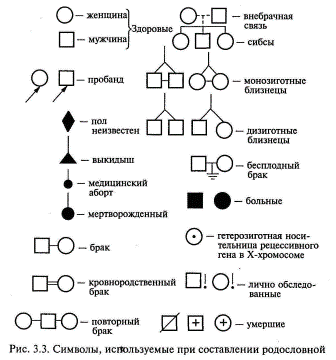
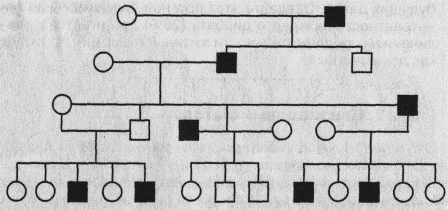
Тема 6. Методы изучения наследственности человекаОсновные закономерности наследственности, установленные для живых организмов, универсальны и в полной мере справедливы и для человека. Вместе с тем, как объект генетических исследований человек имеет свои преимущества и недостатки. Для людей невозможно планировать искусственные браки. Еще в 1923 г. Н. К. Кольцов (1923) отмечал, что «...мы не можем ставить опыты, мы не можем заставить Нежданову выйти замуж за Шаляпина только для того, чтобы посмотреть, какие у них будут дети». Однако эта трудность преодолима, благодаря прицельной выборке из большого числа брачных пар, соответствующих целям данного генетического исследования. Большое число хромосом (46) в значительной мере затрудняет возможности генетического анализа человека. Разработка новейших методов работы с ДНК (метод гибридизации соматических клеток и некоторые другие) устраняют эту трудность. Из-за небольшого числа потомков (во второй половине XX в. в большинстве семей рождалось по 2-3 ребенка) невозможен анализ расщепления в потомстве одной семьи. Однако в больших популяциях можно выбрать семьи с интересующими исследователя признаками. Кроме того, в некоторых семьях определенные признаки прослеживались на протяжении многих поколений, и в таких случаях возможен генетический анализ. Еще одна трудность связана с длительностью смены поколений у человека. Одно поколение занимает в среднем 30 лет, и, следовательно, генетик не может наблюдать более одного-двух поколений. Для человека характерен генотипический и фенотипический полиморфизм. Проявления многих признаков и болезней в значительной степени зависят от условий внешней среды. Следует отметить, что понятие «среда» для человека более широкое, чем для растений и животных. Наряду с питанием, климатом и другими абиотическими и биотическими факторами для человека средой являются и социальные факторы, которые трудно изменяются по желанию исследователя. Вместе с тем, человека как генетический объект широко изучают врачи всех специальностей, что нередко помогает установить различные наследственные отклонения. Итак, генетика человека имеет ряд особенностей: а) на людях запрещены экспериментальные браки; б) рождается малое количество потомков; в) наблюдается позднее половое созревание и большая продолжительность смены поколений (25—30 лет); г) у человека сложный кариотип (много хромосом и групп сцеплений); д) невозможность создания одинаковых условий жизни исследуемых. Несмотря на перечисленные трудности, генетика человека изучена сегодня лучше, чем генетика многих других организмов. В настоящее время интерес и внимание к изучению генетики человека активно возрастают. Так, глобальная международная программа «Геном человека» имеет своей задачей изучение генома человека на молекулярном уровне. Для ее решения используются все новейшие современные методы генетики и медицины. Основные методы изучения наследственности человека: 1. Клииико-генеалогический метод. Он был введен в конце XIX века Ф. Гальтоном и основан на составлении и анализе родословных. Генеалогический метод — это метод изучения родословных, с помощью которого прослеживается распределение болезни (признака) в семье или в роду с указанием типа родственных связей между членами родословной. Эмпирические наблюдения над родословными, в которых отмечалась передача патологических признаков или болезней, известны уже давно. Применение семейного анализа для изучения патологии человека в XVIII—XIX вв. можно рассматривать как предпосылки формирования генеалогического метода. Дальнейшее его усовершенствование шло как по линии составления родословных, так и особенно в отношении методов статистического анализа данных. Генеалогический метод относится к наиболее универсальным методам в медицинской генетике. Этот метод часто называют клинико-генеалогическим, поскольку речь идет об изучении патологических признаков (болезней) в семье с привлечением приемов клинического обследования. Он широко применяется при решении теоретических и прикладных проблем: -для установления наследственного характера признака; -при определении типа наследования признака или заболевания; -для оценки пенетрантности гена; -при анализе сцепления генов и картировании хромосом; -при изучении интенсивности мутационного процесса; -при расшифровке механизмов взаимодействия генов. Особое место генеалогический метод занимает при медико-генетическом консультировании, являясь подчас решающим или единственным методом: -для уточнения природы заболевания; -при постановке диагноза наследственного заболевания; -для дифференциальной диагностики наследственных болезней; -при оценке прогноза заболевания; -при расчете риска для потомства; -для выбора адекватных и оправданных методов дородовой диагностики. При клинико - генеалогическом методе выделяют два последовательных этапа: 1.составление родословной и ее графическое изображение; 2. генетический анализ полученных данных. Подробно рассмотрим каждый из этих этапов. Составление родословной. Сбор сведений о семье начинается с пробанда — индивида, который является предметом интереса врача (исследователя) к данной конкретной родословной. Чаще всего это больной или носитель изучаемого признака. Однако за медико-генетической консультацией могут обращаться и здоровые индивиды по разным причинам, и в этом случае лучше употреблять термин «консультирующийся». Дети одной родительской пары (братья и сестры) называются сибсами. Если сибсы имеют только одного общего родителя, они называются полусибсами. Различают единоутробных (общая мать) и единокровных (общий отец) полусибсов. Обычно родословная собирается по одному или нескольким признакам. Чаще всего больного или консультирующегося беспокоит какое-то конкретное заболевание или признак. Нужно стремиться к наиболее полному составлению родословных по восходящему, нисходящему и боковым направлениям. Эта задача не такая простая, как может показаться на первый взгляд. Чем больше поколений вовлекается в родословную, тем она обширнее. Это может приводить и к искажению получаемой информации, а затем к неточности самой родословной и выводов, сделанных на основе ее анализа. Необходимо собирать сведения, касающиеся не только наличия конкретного заболевания или патологического признака в семье, но и информацию о всех случаях заболеваний, встречающихся среди членов семьи. Важно получить информацию о спонтанных абортах, мертворождениях и ранней детской смертности. Они могут иметь прямое отношение к существу вопроса и сыграть важную роль при оценке прогноза. Всегда нужно получить хотя бы основные данные с обеих сторон (по отцовской и материнской линии), даже если речь идет об аутосомно-доминантном заболевании, унаследованном от одного из родителей. Для анализа и наглядного представления собранной информации используют графическое изображение родословной. Для этого используют стандартные символы (рис. 3.3) и приемы. Однако в зависимости от задач, целей и особенностей родословной составитель может использовать собственные обозначения с обязательным их объяснением под рисунком. Для пояснения принципов обозначения и составления родословных приводим два примера (рис.3.4, 3.5). Как видно из этих рисунков, поколения обозначаются римскими цифрами сверху вниз. Обычно они ставятся слева от родословной. Последнее поколение предков, по которым собрана информация, получает обозначение I поколения. Арабскими цифрами нумеруется потомство одного поколения (весь ряд) слева направо последовательно. Братья и сестры располагаются в родословной в порядке рождения (от старших к младшим). Таким образом, каждый член родословной имеет свой шифр, например, II— 3, III— 5. В тех случаях, когда супруг не исследован на наличие рассматриваемого признака и его родословная не приводится, желательно не изображать его вообще. Все индивиды одного поколения должны располагаться строго в один ряд, поэтому предпочтительно рисовать родословную на разлинованной бумаге. «Подвешивание» символов между рядами поколений является грубой ошибкой. Если родословная очень обширная, то разные поколения располагаются не горизонтальными рядами, а концентрическими.
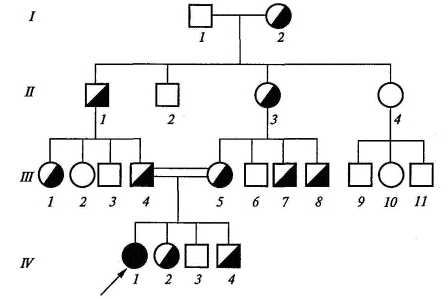
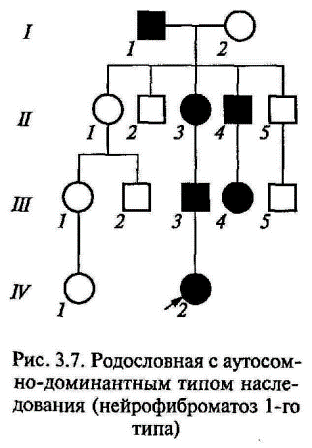
Графическое изображение родословной должно сопровождаться «легендой родословной», которая является обязательным элементом описания родословной и включает: 1) описание состояния здоровья члена родословной, информация о котором важна для понимания характера наследования заболевания (признака) или особенностей его клинического проявления; 2) возраст начала и характер течения заболевания у пораженных; 3) причину смерти и возраст на момент смерти члена родословной; 4) описание методов диагностики и идентификации заболеваний, перечень источников медицинских и других сведений. При применении генеалогического метода важно отмечать в родословной лично обследованных на наличие признака (к этому также можно приравнять получение сведений из объективного источника, например, истории болезни) и необследованных, сведения о которых получены из ответов пробанда или родственников, а также из анкет. Нужно стремиться к получению наиболее полного и объективного первичного материала, который является основой статистического и генетического анализа и, соответственно, залогом правильности и точности полученных в результате выводов. Генеалогический анализ родословной. Целью генеалогического анализа является установление генетических закономерностей. Первая задача при анализе родословной — установление наследственного характера признака. Если в родословной встречается один и тот же признак (или болезнь) несколько раз, можно думать о его наследственной природе. Однако надо прежде всего исключить возможность фенокопии (заболевание протекает сходно с наследственным заболеванием, в то время как причиной его развития является средовой фактор). Например, если патогенный фактор действовал на женщину во время всех ее беременностей, то у нее может родиться несколько детей с одинаковыми врожденными аномалиями. С другой стороны, действие одних и тех же внешних факторов (профессиональных, бытовых и т.д.) на нескольких членов родословной способно также вызывать у них сходные заболевания. Таким образом, если исключается действие сходных внешних факторов, а для представителей разных поколений оно исключается с большей вероятностью, то можно думать о наследственном характере заболевания. После того, как установлен наследственный характер заболевания (признака), необходимо установить тип наследования. Для этого используют принципы генетического анализа и различные статистические методы обработки данных, полученных из родословной. Нетрудно понять, что в большинстве случаев расчеты соотношения числа больных и здоровых в одной конкретной семье могут дать неправильное представление о типе наследования. Это связано, главным образом, со случайным характером наследования различных аллелей. В каждой конкретной семье соотношение больных и здоровых детей может значительно отличаться от теоретически ожидаемых соотношений, характерных для определенного типа наследования. Однако характер родословной, особенности передачи заболевания (признака) в поколениях, соответствие их критериям наследования того или иного типа позволяют сделать определенный вывод о типе наследования патологии (признака) в конкретной семье. Менделевским закономерностям наследования подчиняются только моногенные наследственные заболевания, т. е. заболевания, этиологическим фактором которых является мутация одного гена. В зависимости от локализации и свойств гена различают аутосомно - доминантный и аутосомно-рецессивный типы наследования, когда ген расположен в одной из 22 пар аутосом (неполовых хромосом), Х-сцепленные доминантный и рецессивный типы наследования, когда ген расположен в Х-хромосоме, Y-сцеплен-ное (голандрическое) наследование, когда ген расположен вY-хромосоме, а также митохондриальное (материнское или цитоплазматическое) наследование, когда мутация происходит в геноме митохондрий. 1.1 Гены в семье. Критерии типов наследования Аутосомно-доминантный тип наследования. Если заболевание обусловлено редким аутосомно-доминантным геном, то абсолютное большинство больных в популяции рождаются в браках между пораженным и здоровым супругом. В этом случае один из родителей гетерозиготен по аутосомно-доминантному гену (Аа), а другой гомозиготен по нормальному аллелю (аа). В таком браке возможны следующие варианты генотипов у потомства: Аа, Аа, аа, аа. Таким образом, каждый будущий ребенок независимо от его пола в 50 % случаев имеет вероятность получить от больного родителя как аллель А (и следовательно, быть пораженным), так и нормальный аллель а, и быть здоровым. Таким образом, отношение числа здоровых детей в потомстве к числу пораженных равно 1:1 и не зависит от пола ребенка. В целом основными критериями, позволяющими заподозрить аутосомно-доминантный тип наследования заболевания, являются: заболевание проявляется в каждом поколении без пропусков («вертикальный» тип); каждый ребенок родителя, больного аутосомно-доминантным заболеванием, имеет 50 % - ный риск унаследовать это заболевание; непораженные дети больных родителей свободны от мутантного гена и имеют здоровых детей; заболевания наследуются лицами мужского и женского пола одинаково часто и со сходной клинической картиной. В родословных, представленных на рис. 3.6, 3.7, можно видеть соответствие критериям аутосомно-доминантного типа наследования. Исключения из правила о проявлении аутосомно-доминантных заболеваний в каждом поколении составляют случаи новой мутации (спорадические случаи) или неполной пенетрантности (проявляемости) гена.
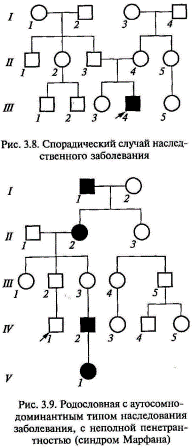
Спорадическим называется единственный случай наследственного доминантного заболевания в семье (рис. 3.8). Он может являться следствием мутации, вновь возникшей в зародышевых клетках одного из здоровых родителей. В случае неполной пенетрантности в родословной с аутосомно-доминантным типом наследования появятся случаи пропуска или «проскока» поколения, т. е. индивид будет иметь пораженного предка и пораженного потомка, а сам при этом будет здоров (рис. 3.9). На сегодняшний день описано около 3000 аутосомно-доминантных признаков человека. Наиболее часто в клинической практике встречаются следующие моногенные заболевания с аутосомно-доминантным типом наследования: семейная гиперхолестеринемия, синдром Марфана, нейрофиброматоз 1-го типа (болезнь Реклингха-узена), синдром Элерса—Данло, миотоническая дистрофия, ахондроплазия, несовершенный остеогенез и др.
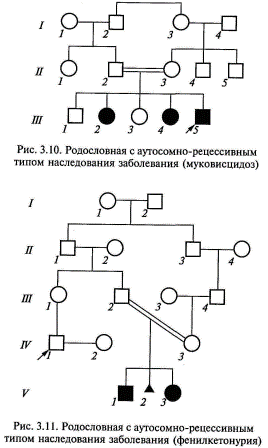
Аутосомно-рецессивный тип наследования. Заболевания с этим типом наследования проявляются только у гомозигот, которые получили по одному рецессивному гену от каждого из родителей. Оно встречается у сибсов пробанда, но иногда встречается и в боковых ветвях родословной. Характерным для аутосомно-рецессивных заболеваний является брак типа Аа х Аа (оба родителя здоровы, но являются носителями мутантного гена). Вероятность рождения больного ребенка в таком браке составляет 25 %. Дети с рецессивными заболеваниями имеют, как правило, фенотипически здоровых родителей, и такие семьи можно определить только после рождения больного ребенка. В популяции встреча двух носителей редкого аутосомно-рецессивного гена — нечастое событие, однако вероятность его значительно возрастает в случае кровного родства супругов. Именно поэтому рецессивные заболевания часто проявляются в кровнородственных браках. Для редких аутосомно-рецессивных заболеваний характерны следующие признаки: родители больного ребенка, как правило, здоровы и являются гетерозиготными носителями патологического аллеля; мальчики и девочки заболевают одинаково часто; повторный риск рождения ребенка с аутосомно-рецессивным заболеванием составляет 25 %; отмечается «горизонтальное» распределение больных, т. е. пациенты чаще встречаются в пределах потомства одной родительской пары; наблюдается увеличение частоты больных детей в родственных браках, причем, чем реже аутосомно-рецессивные заболевания, тем чаще больные происходят из кровнородственных браков; в браке двух пораженных родителей все дети будут больны. Примеры родословных с аутосомно-рецессивным типом наследования приведены на рис. 3.10, 3.11.
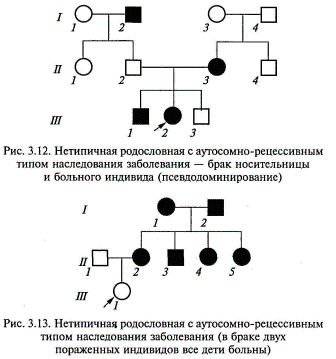
В браке пораженного индивида и гетерозиготного носителя того же мутантного аллеля риск для будущих детей составит 50 %, как при аутосомно-доминантном типе наследования, а не 25%, как должно быть при типичном аутосомно-рецессивном наследовании. Поэтому этот вариант аутосомно-рецессивного наследования носит название псевдодоминирование (рис. 3.12). Родословная, где в браке двух пораженных родителей все дети будут больны, приведена на рис. 3.13. На сегодняшний день известно более 1600 аутосомно-рецессивных заболеваний. По аутосомно-рецессивному типу наследуется абсолютное большинство наследственных заболеваний обмена веществ (ферментопатий). Наиболее частыми и значимыми в клиническом отношении болезнями с аутосомно-рецессивным типом наследования являются: муковисцидоз (кистофиброз поджелудочной железы), фенилкетонурия, адреногенитальный синдром, многие формы нарушения слуха или зрения, мукополисахаридозы, гликогенозы.
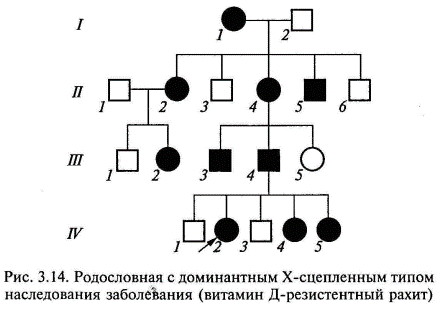
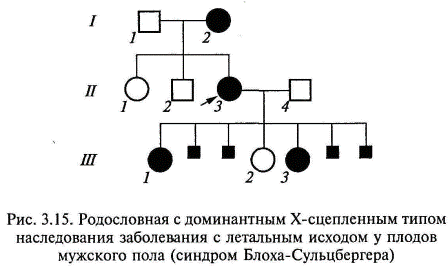
Наследование, сцепленное с Х-хромосомой. Гены, локализованные в половых хромосомах, по-разному распределяются у мужчин и женщин. В клинической генетике практическое значение имеют Х-сцепленные заболевания, т. е. такие, когда патологический ген расположен на Х-хромосоме. Распределение Х-сцепленного признака зависит от распределения Х-хромосомы, несущей аномальный ген. Учитывая то, что у женщин имеются две Х-хромосомы, а у мужчин одна, женщина, унаследовав патологический аллель, будет гетерозиготой, а мужчина — гемизиготой. Этим определяются основные различия в частоте и тяжести поражения различных полов при Х-сцепленном наследовании, как доминантном, так и рецессивном. Доминантный Х-сцепленный тип наследования. Заболевания с Х-сцепленным доминантным типом наследования встречаются в 2 раза чаще у женщин, чем у мужчин. Главная характеристика Х-сцепленного доминантного наследования заключается в том, что больные мужчины передают аномальный ген (или заболевание) всем своим дочерям и не передают его сыновьям. Больная женщина передает Х-сцепленный доминантный ген половине своих детей независимо от пола. Основными признаками Х-сцепленного доминантного типа наследования являются следующие (рис. 3.14): болезнь встречается у мужчин и женщин, но у женщин примерно в два раза чаще; больной мужчина передает мутантный аллель всем своим дочерям и не передает сыновьям, поскольку последние получают от отца Y-хромосому; больные женщины передают мутантный аллель 50 % своих детей независимо от пола; женщины в случае болезни страдают менее тяжело (они гетерозиготы), чем мужчины (являющиеся гемизиготами). Крайней степенью выраженности неравномерности поражения полов при этом типе наследования является существование заболеваний, которыми болеют только индивиды женского пола, в то время как индивиды мужского пола, унаследовавшие патологический аллель, гибнут еще внутриутробно. Подобные редкие заболевания относят к Х-сцепленным доминантным заболеваниям с летальным эффектом у плодов мужского пола (например, синдром недержания пигмента, фокальная кожная гипоплазия) (рис. 3.15).
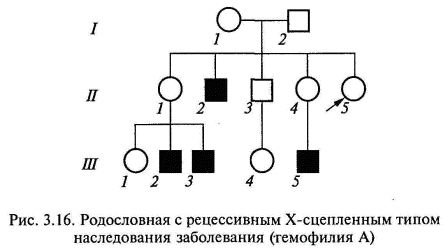
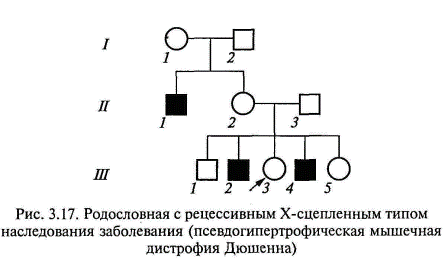
К заболеваниям, характеризующимся Х-сцепленным доминантным наследованием, относятся витамин-Д-резистентный рахит (рахит, не поддающийся лечению обычными дозами витамина Д), ротолицепальцевый синдром и другие болезни. Рецессивный Х-сцепленный тип наследования. Х-сцепленное рецессивное заболевание (или признак) всегда проявляется у мужчин, имеющих соответствующий ген, а у женщин — только в случаях гомозиготного состояния (что наблюдается крайне редко). Основными признаками Х-сцепленного рецессивного наследования являются следующие (рис. 3.16, 3.17): заболевание встречается в основном у лиц мужского пола; признак (заболевание) передается от больного отца через его фенотипически здоровых дочерей половине его внуков; заболевание никогда не передается от отца к сыну; у женщин-носителей иногда выявляются субклинические признаки патологии;
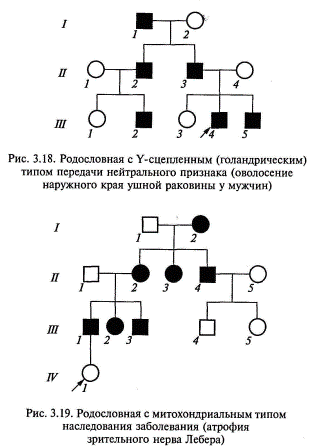
в браке женщины-носительницы с больным мужчиной 50 % дочерей будут больны, 50 % дочерей будут носителями; 50 % сыновей также будут больны, а 50 % сыновей — здоровые (при заболеваниях, не снижающих репродуктивную способность больных мужчин). Выраженные клинические симптомы Х-сцепленных рецессивных заболеваний у женщин могут также наблюдаться в случае сочетания у них этого моногенного дефекта с хромосомной патологией, в частности, с синдромом Шерешевского—Тернера, когда у женщины отсутствует одна из двух Х-хромосом, а также при случайной инактивации Х-хромосомы, несущей нормальный аллель в большинстве клеток организма. У-сцепленное, или голандрическое, наследование. Длительное время полагали, что Y-хромосома содержит только генетически неактивные участки. В настоящее время в Y-хромосоме выявлена локализация около 20 генов, в том числе гены, определяющие развитие семенников, отвечающие за сперматогенез, контролирующие интенсивность роста, определяющие оволосение ушной раковины, средних фаланг кистей и некоторые другие. Признак, гены которого локализованы в Y-хромосоме, передается от отца всем мальчикам и только мальчикам. Патологические мутации, обусловливающие нарушения формирования семенников или сперматогенеза, не наследуются в связи со стерильностью их носителей. Пример родословной с У-сцепленным типом наследования (оволосение ушной раковины) представлен на рис. 3.18. „- Митохондриальная или цитоплазматическая наследственность. Митохондриальный геном представлен в виде кольцевой молекулы ДНК, содержащей 16 569 тыс. пар оснований. На сегодняшний день известен целый ряд мутаций митохондриальной ДНК, вызывающий различные заболевания. Поскольку митохондрии наследуются ребенком от матери с цитоплазмой ооцитов (в зрелых спермиях присутствуют только
4 митохондрии), то митохондриальный тип наследования будут характеризовать два основных признака (рис. 3.19): заболевание передается только от матери всем детям независимо от пола ребенка; больные отцы не передают заболевание ни сыновьям, ни дочерям — все дети будут здоровыми и передача заболевания прекращается. Согласно этому типу наследуются атрофия зрительного нерва Лебера, митохондриальная миоэнцефалопатия, синдром Кернса— Сейра и некоторые другие заболевания. В настоящее время описано около 30 различных заболеваний, при которых обнаружены мутации митохондриальной ДНК. Гены митохондрий кодируют различные ферменты дыхательной цепи в клетках. Таким образом, они принимают участие в энергетических процессах, где выполняют довольно универсальные функции и экспрессируются во многих тканях, особенно в нервной и мышечной системе, чем и обусловливается многообразие клинических проявлений митохондриальных наследственных болезней: миопатия, судороги, офтальмоплегия, нарушение сердечного ритма, а также поражение печени и почек. Контрольные вопросы и задания 1. Какие вопросы можно решить с помощью клинико - генеалогического метода? 2. Перечислите этапы клинико-генеалогического метода. 3. Что означают термины «пробанд», «сибсы», «родственный брак»? 4. Перечислите критерии аутосомно-доминантного типа наследования и риведите примеры заболеваний. 5. Что такое спорадический случай? 6. Перечислите критерии аутосомно-рецессивного типа наследования и назовите заболевания, наследуемые по этому типу. 7. Что понимают под псевдодоминированием? 8. Охарактеризуйте различия между Х-сцепленным доминантным и Х-сцепленным рецессивным типами наследования? 9. Чем характеризуется митохондриальный тип наследования? 10. Каковы критерии голандрического типа наследования? 11. Перечислите основные принципы составления родословной, типы родословных схем, условные обозначения. 12. Перечислите особенности: а) аутосомно-доминантного наследования; б) аутосомно-рецессивного наследования; в) наследования, сцепленного с полом. 13. Определите характер наследования (доминантный или рецессивный) аутосомного признака, запишите генотипы всех членов родословной, приведенной ниже.
14. Известно, что подагра наследуется по аутосомно-доминантному типу. По некоторым данным, пенетрантность этого гена у мужчин составляет 20%, а у женщин она равна 0. Какова частота заболевания подагрой в семье, где оба родителя гетерозиготны? Какова вероятность заболевания подагрой детей в случае, где один из родителей гетерозиготен, а другой гомозиготен по анализируемому признаку? 15. Определите тип наследования аутосомного признака (доминантный или рецессивный), укажите генотипы всех членов родословной:
16. Мужчина, страдающий дальтонизмом и глухотой, женился на женщине, нормальной по зрению и хорошо слышащей. У них родились — глухой сын-дальтоник и дочь-дальтоник с нормальным слухом. Определите вероятность рождения в этой семье дочери с обеими аномалиями, если известно, что дальтонизм и глухота передаются как рецессивные признаки, но дальтонизм сцеплен с Х-хромосомой, а глухота — аутосомный признак. 17. Гипертрихоз (избыточное оволоснение) передается через Y-хромосому, а полидактилия (шестипалость) — доминантный аутосомный признак. В семье, где отец имел гипертрихоз, а мать — полидактилию, родилась дочь, нормальная в отношении обоих признаков. Какова вероятность того, что следующий ребенок в этой семье будет также без обеих аномалий? 18. Женщина-правша с карими глазами и нормальной кровью вышла замуж за голубоглазого мужчину-правшу, страдающего гемофилией. У них родилась голубоглазая дочь-левша с гемофилией. Какова вероятность того, что следующий ребенок в этой семье будет левшой, страдающим гемофилией? Известно, что карий цвет глаз и праворукость — доминантные признаки, а гемофилия — рецессивный, сцепленный с Х-хромосомой признак. Какой цвет глаз возможен у больных детей? 19. Арахнодактилия (паучья кисть) наследуется как доминантный аутосомный признак с пенетрантностью 30%. Леворукость — рецессивный аутосомный признак с полной пенетрантностью. Определите вероятность рождения ребенка с двумя аномалиями в семье, в которой оба родителя гетерозиготны по обоим парам признаков. 10. Проведите анализ и определите характер наследования признака по родословной, представленной ниже:
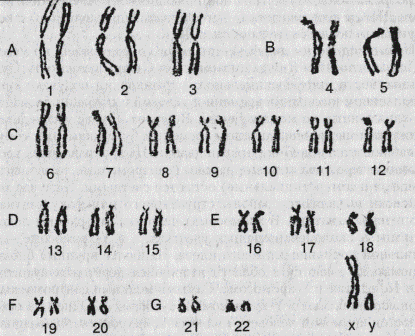
Признак сцеплен с полом. В какой из половых хромосом локализован ген? Укажите его доминантность или рецессивность. 20. Пробанд страдает ночной слепотой. Его два брата тоже больны. По линии отца пробанда страдающих ночной слепотой не было. Мать пробанда больна. Две ее сестры и два брата здоровы; их дети также здоровы. Бабушка по материнской была больна, дедушка здоров; сестра бабушки больна, а брат здоров. Прадедушка (отец бабушки), его сестра и брат, а также прапрадедушка и его брат, имевший больную дочь и двух больных сыновей, также были больны. Жена пробанда, ее родители и родственники здоровы. Определите вероятность рождения больных детей в семье пробанда. 21. У супругов с нормальным зрением четверо детей: две дочери и два сына. У первой дочери зрение нормальное; у нее три сына, два из которых дальтоники. У второй дочери и пяти ее сыновей зрение нормальное. Первый сын страдает дальтонизмом, а его две дочери и два сына видят нормально. Второй сын и четверо его сыновей имеют нормальное зрение. Каковы генотипы бабушки, дедушки, их детей и внуков? Составьте родословную данной семьи. 22. Пенетрантность по шизофрении у гетерозигот составляет 20%, у гомозигот — 100%. Мужчина, страдающий периодическими обострениями шизофрении, женится здоровой женщине. Известно, что у родственников жены такой патологии не было. Бабушка мужа была больна, но его родители здоровы. Каков прогноз для этой семьи? 14. Здоровая девушка в возрасте 22 лет, один из родителей которой болен сахарным диабетом, обращается в генетическую консультацию за прогнозом по поводу себя и своих будущих детей. Сделайте этот прогноз. Максимальная пенетрантность сахарного диабета (20%) достигается с увеличением продолжительности жизни. Рассмотрите признак как доминантный. 2 Цитогенетический метод. Основными показаниями для цитогенетического исследования являются: 1) пренатальная диагностика пола плода в семьях, отягощенных заболеваниями, сцепленными с Х-хромосомой; 2) недифференцированная олигофрения (слабоумие); 3) привычные выкидыши и мертворождения; 4) множественные врожденные пороки развития у ребенка; 5) бесплодие у мужчин; 6) нарушение менструального цикла (первичная аменорея); 7) пренатальная диагностика при возрасте матери старше 35 лет. Материалом для патогенетического исследования могут быть: клетки периферической крови (лимфоциты); фибробласты кожи; клетки, полученные при амниоцентезе или биопсии хориона; клетки абортусов, мертворожденных и др. Основа метода — микроскопическое изучение хромосом человека. Цитогенетические исследования стали широко использоваться с начала 20-х гг. XX в. для изучения морфологии и подсчета хромосом человека, культивирования лейкоцитов для получения метафазных пластинок. Развитие современной цитогенетики человека связано с именами цитологов Д. Тио и А. Левана. В 1956 г. они первыми установили, что у человека 46, а не 48, как думали раньше, хромосом. Это событие положило начало широкому изучению митотических и мейотических хромосом человека. В 1959 г. французские ученые Д. Лежен, Р. Тюрпен и М. Готье установили хромосомную природу болезни Дауна. В последующие годы были описаны многие часто встречающиеся у человека хромосомные болезни. Цитогенетика стала важнейшим разделом практической медицины. В настоящее время ци тогенетический метод применяется для диагностики хромосомных болезней, составления генетических карт хромосом, изучения мутационного процесса и др. В 1960 г. в Денвере (США) была разработана первая Международная классификация хромосом человека. В ее основу легли размеры хромосом и положение первичной перетяжки — центромеры. Все хромосомы по форме разделены на метацентрические, субметацентрические и акроцентрические и подразделены на 7 групп, обозначенных латинскими буквами А, В, С, В, Е, F и G. Каждая пара хромосома обозначена порядковым номером от 1 до 23, отдельно выделены половые хромосомы — X и Y (рис. 3) У женщин две Х-хромосомы, у мужчин X и Y-хромосомы. Х-хромосома у женщин не отличается от аутосом группы С; Y-хромосома акроцентрическая, сходная с хромосомами группы С, не имеет спутников. Длина короткого плеча может значительно изменяться. Аутосомы группы С и В содержат в коротких плечах районы ядрышкового организатора. В 1971 г. на I Пражской конференции генетиков в дополнении к Денверской классификации были представлены методы дифференциальной окраски хромосом, благодаря которым каждая хромосома приобретает свой неповторимый рисунок, что помогает точной идентификации. Основные сведения о морфологии хромосом человека получены при изучении их в метафазах митоза и профазе-метафазе мейоза. При этом важно, чтобы количество делящихся клеток было достаточно высоко. Важнейшие цитогенетические работы выполнены на лимфоцитах периферической крови, поскольку культивирование лимфоцитов в течение 2—3 суток в присутствии фитогемагглютинина позволяет получить множество метафазных пластинок для хромосомного анализа. Цитогенетическому анализу подвергают однослойные метафазные пластинки с раздельно лежащими хромосомами. Для этого делящиеся клетки обрабатывают колхицином и некоторыми другими химическими веществами (гипотоническим солевым раствором, метанол-уксусным фиксатором и др.). Важным этапом цитогенетического анализа является окраска полученных препаратов. Ее проводят простыми, дифференциальными и флюоресцентными методами. Простая окраска обеспечивает групповую идентификацию хромосом. Используется она для количественного учета хромосомных аномалий при определении мутагенности среды (действия радиации, химических мутагенов и др.). С помощью этого типа окраски были открыты многие хромосомные болезни, а также хромосомные аберрации, вызывающие самопроизвольные аборты, врожденные пороки развития, канцерогенез и т. п. В 70-е гг. XX в. в медицинской практике начали применяться методы дифференциального окрашивания, выявляющие структурную разнородность хромосом по длине, что выражается в виде чередования светлых и темных полос (эу- и гетерохроматических районов). Отмечается, что протяженность и рисунок полос специфичны для каждой хромосомы. Дифференциальное окрашивание хромосом можно проводить радом способов. Первоначально использовали акрихинипритфлюоресцентное алкилирующее вещество (Ц-метод).
Действие его основано на способности метафазных хромосом дифференциально связывать флюорохромы. После окрашивания акрихином сегменты приобретают яркое флюоресцирующее свечение. Рисунок каждой хромосомы специфичен по числу, размерам и положению по-разному флюоресцирующих сегментов, что и обеспечивает идентификацию всех хромосом. С помощью данного метода окраски можно идентифицировать хроматин с повышенным содержанием АТ-пар, поскольку они активнее флюоресцируют. Специфическим преимуществом Q - метода является то, что он позволяет даже в интерфазном ядре идентифицировать Y-хромосому по яркому свечению. Для просмотра таких препаратов используют люминесцентный микроскоп. В дальнейшем был разработан способ окраски хромосом без флюоресцентных красителей — G-окраска (краситель Гимза). После предварительной инкубации в солевом растворе хромосомы обрабатываются протеазой. В результате хромосомы приобретают сегментированный вид благодаря чередованию темно- и светлоокрашенных участков. Механизм образования сегментов пока недостаточно ясен. Предполагается, что окрашенные сегменты — это гетерохроматиновые участки с повторяющимися последовательностями ДНК, а неокрашенные — это эухроматиновые районы с кодирующими последовательностями ДНК. К разновидностям дифференциального окрашивания по методу Гимзы относятся R-окрашиваемость и C-окрашиваемость. Эти разновидности дифференциального окрашивания получают при определенном изменении времени и условий инкубации препаратов, окрашенных по методу Гимзы. В первом случае распределение окрашенных и неокрашенных сегментов будет обратным тому, что наблюдается при G- и Q - окрашивании. На R-окрашенных хромосомах гетерохроматиновые районы (центромерные, околоцентромерные и интерстициальные) остаются светлыми. В случае же С-окраски выявляются районы структурного или факультативного гетерохроматина. В хромосомах человека эти районы локализованы в околоцентромерных участках, а в Y-хромосоме — в дистальной половине длинного плеча. Наиболее крупные блоки С-хроматина имеются в области вторичных перетяжек аутосом 1,9 и 16, а также в Y-хромосоме. Самыми мелкими центромерными блоками обладают Y-хромосома и аутосома 2. Одной из особенностей хромосом человека является асинхронность (неодновременность) репликации по длине. В каждой хромосоме есть рано и поздно реплицирующиеся участки. Для выявления последовательности репликации применяется 5-бромдезоксиуридин — аналог тимина. Включившие его участки окрашиваются слабо. Применяется 5-бром-дезоксиуридин и для дифференциальной окраски сестринских хроматид, если он вводится на полный клеточный цикл. В этом случае вновь образуемая хроматида включит этот аналог тимина и будет окрашена слабо, а другая (старая) окрасится интенсивно. Этот метод позволяет выявлять участки сестринских хроматидных обменов (СХО). При воздействии различными мутагенными факторами число СХО увеличивается, следовательно, этот метод выгоден для изучения мутационного процесса у человека. Успехи молекулярной цитогенетики человека позволяют разрабатывать новые методы изучения хромосом. Так, следует отметить метод флюоресцентной гибридизации in situ (FISH-метод), который дает FISH возможность исследовать широкий круг вопросов от локализации гена до расшифровки сложных перестроек между несколькими хромосомами. Метод может применяться и для диагностики анеуплоидий в интерфазных ядрах. Таким образом, соединение цитогенетических и молекулярно-генетических методов в генетике человека делает почти неограниченными возможности диагностики хромосомных аномалий. Вопросы и задания 1. Дайте определение следующим терминам: хромосома, хроматида, хроматин, хромомера, кариотип. 2. В чем различие между эухроматином и гетерохроматином? Типы хроматина. Ответ обоснуйте. 3. На каких стадиях митоза хромосомы хорошо видны? Почему? 4. Дайте характеристику нормального кариотипа в соответствии с Денверской классификацией. 5. Опишите методы дифференциальной окраски хромосом и их роль в развитии цитогенетики человека. 6. Какое практическое значение имеет исследование полового хроматина 3.Генетика соматических клеток. Она изучает наследственность и изменчивость соматических клеток. Благодаря тому, что эти клетки содержат весь объем генетической информации, на них можно также изучать генетические особенности целостного организма. Генетика соматических клеток позволила включить человека в группу экспериментальных объектов. Соматические клетки человека для генетических исследований получают из материала биопсий (прижизненное иссечение тканей или органов) и аутопсий (кусочки тканей или органов от трупов). Чаще всего используют клеточные культуры фибробластов и лимфоидных клеток. В настоящее время применяют следующие методы генетики соматических (клеток человека: 1) простое культивирование; 2) гибридизация; 3) клонирование; 4) селекция. П р о с т о е к у л ь т и в и р о в а н и е - - размножение клеток на питательных средах с целью получения их в достаточном количестве для цитогенетического, биохимического, иммунологического и других методов исследования. Гибридизация соматических клеток - это слияние клеток двух разных типов. Гибридизацию могут проводить между клетками, полученными от разных людей, а также клетками человека с клетками мыши, крысы, китайского хомячка, морской свинки, обезьяны, курицы. Спонтанное (произвольное) слияние происходит редко, поэтому в смешанную культуру добавляют или чаще вирус Сендай. При слиянии клеток образуется гетерокарион (гибридная клетка с двумя ядрами разных тиков клеток). Затем ядра этой клетки могут слиться с образованием синкариона (от греч. syn — вместе). Особый интерес представляют гибридные клетки «человек—мышь», так как при последующих делениях они имеют тенденцию к утрате многих хромосом человека. Примерно через 30 поколений можно найти клетки, содержащие только одну—две пары человеческих хромосом. Если в гибридной клетке отсутствует какая-либо хромосома и не происходит синтез каких-то белков, то можно предположить, что гены, детерминирующие синтез этих белков, локализованы в данной хромосоме. Этот метод позволяет установить группы сцепления, а используя хромосомные перестройки (нехватки и транслокации), выяснять последовательность расположения генов и строить генетические карты хромосом человека. 4.Клонирование- получение потомков одной клетки (клона), взятой из общей клеточной массы. Все клетки будут с одинаковым генотипом. Одним из примеров метода клонирования является получение гибридом (от лат. Hibrida- — помесь и греч. oma — опухоль). Гибридома — это клеточный гибрид, получаемый слиянием нормального лимфоцита и опухолевой клетки Селекция (от лат. selectio — отбор, выбор) — отбор клеток с заранее заданными свойствами при культивировании их на селективных питательных средах. Например, если использовать питательную среду без лактозы (но с добавлением других cахаров), то из большого числа клеток помещенных в нее, может оказаться несколько, способных существовать без лактозы. В дальнейшем можно будет получить клон этих клеток. 5.Метод дерматоглифики. Представляет собой изучение папиллярных узоров пальцев, ладоней и стоп для определения зиготности близнецов, диагностики некоторых геномных и хромосомных мутаций (например, болезни Дауна, Патау и других); для идентификации личности в криминалистике, установления отцовства в судебной медицине. 6.Близнецовый метод. Это метод изучения генетических закономерностей на близнецах. Он позволяет оценить относительную роль (удельный вес) генетических и средовых факторов в развитии конкретного признака или заболевания. Впервые он был предложен Ф. Гальюном в 1875 г. Близнецовый метод дает возможность определить вклад генетических (наследственных) и средовых факторов (климат, питание, обучение, воспитание и др.) в развитии конкретных признаков или заболеваний у человека. При использовании близнецового метода проводится сравнение: монозиготных (однояйцовых) близнецов с дизиготными; партнеров в монозиготных парах между собой; данных анализа близнецовой выборки с общей популяцией. Монозиготные близнецы (МБ) образуются из одной зиготы, разделившейся на стадии дробления на две (или более) части. С генетической точки зрения они идентичны, т. е. обладают одинаковыми генотипами. Монозиготные близнецы всегда одного пола (рис. 2). Особую группу среди МБ составляют необычные типы близнецов: двухголовые (как правило, нежизнеспособные), каспофаги («сиамские близнецы»). Родившиеся в 1811 г. в Сиаме (ныне Таиланд) сиамские близнецы Чанг и Энг прожили 63 года. Они были женаты на близнецах; Чанг произвел на свет 10, а Энг — 12 детей. От бронхита умер Чанг, а спустя 2 часа умер и Энг. Их связывала тканевая перемычка шириной около 10 см. Позднее было установлено, что эта перемычка состояла из печеночной ткани и связывала две печени. Любая хирургическая попытка разделить братьев в то время вряд ли была бы успешной. В настоящее время разъединяют и более сложные связи между близнецами. Дизиготные близнецы (ДБ) развиваются в случае, если образуются одновременно две яйцеклетки, оплодотворенные двумя сперматозоидами.
Естественно, что дизиготные близнецы имеют различные генотипы. Они сходны между собой не более, чем братья и сестры, т. к. имеют около 50% идентичных генов. Общая частота рождения близнецов составляет примерно 1%; из них около 1/3 приходится на монозиготных близнецов. Известно, что число рождений монозиготных близнецов сходно в разных популяциях, в то время как для дизиготных близнецов эта цифра существенно различается. Например, в США дизиготные близнецы рождаются чаще среди негров, чем белых. В Европе частота появления дизиготных близнецов составляет 8 на 1000 рождений. Однако в отдельных популяциях их бывает больше. Самая низкая частота рождения близнецов, присущая в большей степени монголоидным популяциям, наблюдается в Японии. Отмечается, что частота врожденных уродств у близнецов, как правило, выше, чем у одиночнорожденных. Полагают, что многоплодие генетически обусловлено. Однако это справедливо лишь для дизиготных близнецов. Факторы, влияющие на частоту рождения близнецов, в настоящее время мало изучены. Есть данные, показывающие, что вероятность рождения дизиготных близнецов повышается с увеличением возраста матери, а также порядкового номера рождения. Влияние возраста матери объясняется, вероятно, повышением уровня гонадотропина, приводящее к учащению полиовуляции. Имеются данные о снижении частоты рождения близнецов в индустриальных странах. Близнецовый метод включает в себя диагностику зиготности близнецов. В настоящее время используются следующие методы для ее установления: 1) полисимптомный метод заключается в сравнении пары близнецов по внешним признакам (форма бровей, носа, губ, ушных раковин, цвет волос, глаз и т. п.). Несмотря на очевидное удобство, этот метод до известной степени субъективен и может давать ошибки; 2) иммуногенетический метод более сложен и основывается на анализе групп крови, белков сыворотки крови, лейкоцитарных антигенов, чувствительности к фенилтиокарбамиду и др. Если у близнецов по этим признакам различий нет, их считают монозиготными; 3) достоверным критерием зиготности близнецов является приживляемость кусочков кожи. Установлено, что у дизиготных близнецов такая пересадка всегда заканчивается отторжением, в то время как у монозиготных пар отмечается высокая приживляемость трансплантантов; 4) метод дерматоглифики заключается в изучении папиллярных узоров пальцев, ладоней и стоп. Эти признаки строго индивидуальны и не изменяются в течение всей жизни человека. Не случайно, что эти показатели используются в криминалистике и судебной медицине для опознания личности и установления отцовства. Сходство дерматоглифических показателей у монозиготных близнецов значительно выше, чем у дизиготных. Близнецовый метод включает также сопоставление групп моно-и дизиготных близнецов по изучаемому признаку. Если какой-либо признак встречается у обоих близнецов одной пары, то она называется конкордантной, если же у одного из них, то пара близнецов называется дискордантной (конкордантность — степень сходства, дискордантность — степень различия). При сопоставлении моно- и дизиготных близнецов определяют коэффициент парной конкордантности, указывающий на долю близнецовых пар, в которых изучаемый признак проявился у обоих партнеров. Коэффициент конкордантности выражается в долях единицы или в процентах и определяется по формуле: K=C/(C+D) где С — число конкордатных пар Д — число дискордантных пар. Сравнение парной конкордантности у моно- и дизиготных близнецов дает ответ о соотносительной роли наследственности и среды в развитии того или иного признака или болезни. При этом исходят из предположения, что степень конкордантности достоверно выше у монозиготных, чем у дизиготных близнецов, если наследственные факторы имеют доминирующую роль в развитии признака (см. табл. 1). Таблица 1. Конкордантность некоторых признаков человека у однояйцовых (ОБ) и двуяйцовых (ДБ) близнецов
Если значение коэффициента конкордантности примерно близко у монозиготных и дизиготных близнецов, то считают, что развитие признака определяется, главным образом, негенетическими факторами, т. е. условиями среды. Если в развитии изучаемого признака участвуют как генетические, так и негенетические факторы, то у монозиготных близнецов будут иметь место определенные внутрипарные различия. При этом будут уменьшаться различия между моно- и дизиготными близнецами по степени конкордантности. В этом случае считают, что к развитию признака имеется наследственная предрасположенность. Для количественной оценки роли наследственности и среды в развитии того или иного признака используют различные формулы. Чаще всего пользуются коэффициентом наследуемости, который вычисляется по формулам: Н = КМБ - КДБ: 100 - КДБ (в процентах); Н = КМБ — КДБ: 1 — КДБ (в долях единицы), где Н — коэффициент наследуемости, К — коэффициент парной конкордантности в группе монозиготных или дизиготных близнецов. В зависимости от значения Н судят о влиянии генетических и средовых факторов на развитие признака. Например, если значение Н близко к нулю, считают, что развитие признака обусловлено только факторами внешней среды. При значении Н от 1 до 0,7 — наследственные факторы имеют доминирующее значение в развитии признака или болезни; среднее значение Н от 0,4 до 0,7 свидетельствует о том, что признак развивается под действием факторов внешней среды при наличии генетической предрасположенности. Рассмотрим несколько примеров. Как уже было отмечено, группы крови у человека полностью обусловлены генотипом и не изменяются под влиянием среды. Коэффициент наследуемости равен 100%. По некоторым морфологическим признакам (форме носа, бровей, губ и ушей, цвету глаз, волос и кожи) монозиготные близнецы конкордантны в 97—100%, а дизиготные (в зависимости от признака) — в 70—20% случаев. Конкордантность МБ по заболеваемости шизофренией равна 70%, а у ДБ — 13%. Тогда Н = (70 - 13): (100 - 13) - 0,65 или 65%. В данном случае преобладают генетические факторы, но существенную роль играют и условия среды. С помощью близнецового метода выявлено значение генотипа и среды в патогенезе многих инфекционных болезней. Так, при заболевании корью и коклюшем ведущее значение имеют инфекционные факторы, а при туберкулезной инфекции существенное влияние оказывает генотип. Исследования, проводимые на близнецах, помогут ответить на такие вопросы, как влияние наследственных и средовых факторов на продолжительность жизни человека, развитие одаренности, чувствительность к лекарственным препаратам и др. В настоящее время близнецовый метод в генетике человека используется в сочетании с другими методами генетического анализа. Вопросы и задания 1. В чем отличия между монозиготными и дизиготными близнецами? 2. Приведите основные методы диагностики моно- и дизи-готности. 3. Примените близнецовый метод для анализа альтернативных признаков. 4. Приведите примеры конкордантности близнецов при некоторых инфекционных и полифакториальных заболеваниях. 7.Биохимические методы. Они позволяют выявить изменения в обмене веществ для уточнения диагноза заболевания, гетерозиготного носительства. Например, гетерозиготные носители рецессивного аллеля фенилкетонурии реагируют на введение фенилаланина более сильным повышением концентрации аминокислоты в плазме, чем нормальные гомозиготы. Этот метод используют в медико-генетическом консультировании для определения вероятности рождения ребенка с наследственным заболеванием. Заболевания, в основе которых лежит нарушение обмена веществ, составляют значительную часть наследственной патологии (фенилкетонурия, галактоземия, алкаптонурия и др.). Предположить наличие у больного наследственного дефекта обмена можно по следующим признакам: 1) умственная отсталость, изолированная или в сочетании с патологией других органов; 2) нарушение психического статуса; 3) нарушение физического развития; 4) судороги, мышечная гипо- или гипертония, нарушение походки и координации движений, желтуха, гипо- или гиперпигментация; 5) непереносимость отдельных пищевых продуктов и лекарственных препаратов, нарушение пищеварения и др. 8.Популяционно-статистический метод. Одним из важных в современной генетике направлений является популяционная генетика. Она изучает генетическую структуру популяций, их генофонд, взаимодействие факторов, обусловливающих постоянство и изменение генетической структуры популяций. Под популяцией в генетике понимается совокупность свободно скрещивающихся особей одного вида, занимающих определенный ариал и обладающих общим генофондом в ряду поколений. Генофонд — это вся совокупность генов, встречающихся у особей данной популяции. В медицинской генетике популяционно-статистический метод используется при изучении наследственных болезней населения, частоты нормальных и патологических генов, генотипов и фенотипов в популяциях различных местностей, стран и городов. Кроме того, этот метод изучает закономерности распространения наследственных болезней в разных по строению популяциях и возможность прогнозировать их частоту в последующих поколениях. Популяционно-статистический метод используется для изучения: — частоты генов в популяции, включая частоту наследственных болезней; — закономерности мутационного процесса; — роли наследственности и среды в возникновении болезней с наследственной предрасположенностью; — влияния наследственных и средовых факторов в создании фенотипического полиморфизма человека по многим признакам и др. Использование популяционно-статистического метода включает правильный выбор популяции, сбор материала и статистический анализ полученных результатов. В основе метода лежит закономерность, установленная в 1908 г. английским математиком Дж. Харди и немецким врачом В. Вайнбергом для идеальной популяции. Обнаруженная ими закономерность получила название закона Харди— Вайнберга. Для идеальной популяции характерны следующие особенности: большая численность популяции, свободное скрещивание (панмиксия) организмов, отсутствие отбора и мутационного процесса, отсутствие миграций в популяцию и из нее. В идеальной популяции соотношение частоты доминантных гомозигот (АА), гетерозигот (Аа) и рецессивных гомозигот (аа) сохраняется постоянным из поколения в поколение, если никакие эволюционные факторы не нарушают это равновесие. В этом основной смысл закона Харди— Вайнберга. При изменении любого из этих условий равновесия соотношение численности генотипов в популяции нарушается. К этим условиям относятся родственные браки, мутации, дрейф генов, отбор, миграции и другие факторы. Однако это не снижает значения закона Харди— Вайнберга. Он является основой при рассмотрении генетических преобразований, происходящих в естественных и искусственно созданных популяциях растений, животных и человека. Соотношение численности разных генотипов и фенотипов в панмиктической популяции определяется по формуле бинома Ньютона: (p+q)2=p2+2pq+q2; (p+q)2=1 где р — частота доминантного аллеля А; q — частота рецессивного аллеля а; р2 — частота генотипа АА (гомозигот по доминантному аллелю); q2 — частота генотипа аа (гомозиготы по рецессивному аллелю). В соответствии с законом Харди— Вайнберга частота доминантных гомозигот (АА) равна квадрату вероятности встречаемости доминантного аллеля, частота гетерозигот (Аа) — удвоенному произведению вероятности встречаемости доминантного и рецессивного аллелей. Частота встречаемости рецессивных гомозигот (аа) равна квадрату вероятности рецессивного аллеля. Таким образом, популяционно-статистический метод дает возможность рассчитать в популяции человека частоту нормальных и патологических генов-гетерозигот, доминантных и рецессивных гомозигот, а также частоту нормальных и патологических фенотипов, т. е. определить генетическую структуру популяции. Важным фактором, влияющим на частоту аллелей в малочисленных популяциях и в изолятах, являются генетико-автоматические процессы, или дрейф генов. Это явление было описано в 30-х гг. Н. П. Дубининым и Д. Д. Ромашевым (СССР), С. Райтом и Р. Фишером (США). Оно выражается в случайных изменениях - частоты аллелей, не связанных с их селективной ценностью и действием естественного отбора. В результате дрейфа генов адаптивные аллели могут быть элиминированы из популяции, а менее адаптивные и даже патологические (в силу случайных причин) могут сохраниться и достигнуть высоких концентраций. В результате в популяции может происходить быстрое и резкое возрастание частот редких аллелей. Генетико-автоматические процессы наиболее интенсивно протекают при неравномерном размножении особей в популяции. Колебания численности популяции нередко наблюдается у насекомых, грызунов и других животных в виде так называемых «волн жизни». В отдельные благоприятные годы численность их сильно возрастает, а затем резко падает. Причинами могут быть развитие заболеваний, нехватка пищи, понижение температуры и др. В результате спада численности популяции или в изолированных популяциях уменьшается гетерозиготность и возрастает генетическая однородность популяции. Примером действия дрейфа генов в человеческих популяциях может служить «эффект родоначальника». Он наблюдается, если структура популяции формируется под влиянием аллелей ограниченного числа семей. В таких популяциях нередко наблюдается высокая частота аномального гена, сохранившегося в результате случайного дрейфа генов. Возможно, что следствием дрейфа генов является разная частота резус-отрицательных людей в Европе (14%) и в Японии (1%), неравномерное распространение наследственных болезней по разным группам населения земного шара. Например, в некоторых популяциях Швеции широко распространен ген юве-нильной амавротической идиотии, в Южной Африке — ген порфи-рии, в Швейцарии — ген наследственной глухоты и др. Близкородственные браки (инбридинг) значительно влияют на генотипический состав популяции. Такие браки чаще всего заключаются между племянницей и дядей, двоюродными братом и сестрой. Близкородственные браки запрещены во многих странах. Это связано с высокой вероятностью рождения детей с наследственной патологией. Родственники, имея общее происхождение, могут быть носителями одного и того же рецессивного патологического гена, и при браке двух здоровых гетерозигот вероятность рождения больного ребенка становится высокой. Новые гены могут поступать в популяцию в результате миграции (потока генов), когда особи из одной популяции перемещаются в другую и скрещиваются с представителями данной популяции. Реальные популяции редко бывают полностью изолированными. Всегда происходит некоторое передвижение особей из одной популяции в другую. Оно может быть не только активным, но и пассивным (перенос семян птицами). Иногда человек умышленно перемешивает популяции. Например, в Сибири для улучшения местных соболей в их популяции выпускают баргузинских соболей с очень темной окраской меха, более ценимой в меховой промышленности. Это приводит к изменению частоты аллелей в основной популяции и среди «иммигрантов». В локальных популяциях частота аллелей может изменяться, если у старожилов и пришельцев исходные частоты аллелей различны. Аналогичные процессы происходят и в человеческих популяциях. В США потомство от смешанных браков между белыми и неграми относится к негритянскому населению. По данным Ф. Айала и Дж. Кайгера (1988) частота аллеля, контролирующего резус-фактор у белого населения, составляет 0,028. В африканских племенах, от которых происходит современное негритянское население, частота этого аллеля равна 0,630. Предки современных негров США были вывезены из Африки 300 лет назад (около 10 поколений). Частота аллеля у современного негритянского населения Америки составляет 0,446. Таким образом, поток генов от белого населения к негритянскому шел со скоростью 3,6% за 1 поколение. В результате через 10 поколений доля генов африканских предков составляет сейчас 0,694 общего числа генов современного негритянского населения США. Около 30% генов американские негры унаследовали от белого населения. Очевидно, поток генов между белым и негритянским населением был значительным. Наконец, следует кратко рассмотреть, как влияют на генетическую структуру популяций мутационный процесс и отбор. Мутации как фактор эволюции обеспечивают приток новых аллелей в популяции. По изменению генотипа мутации подразделяют на генные (или точковые), внутрихромосомные и межхромосомные, геномные (изменение числа хромосом.). Генные мутации могут быть прямыми (А а) и обратными (а А). Частота возникновения прямых мутаций значительно выше обратных. Одни и те же гены могут мутировать многократно. Кроме того, один и тот же ген может изменяться в несколько аллельных состояний, образуя серию множественных аллелей (А а1, а2, а3, аn). Изучение частоты мутаций, обусловливающих у человека такие тяжелые болезни, как гемофилия, ретинобластома, пигментная ксеродерма и др., дает основание полагать, что частота возникновения патологических мутаций отдельного гена составляет около 1 —2 на 100 тыс. гамет за поколение. Учитывая общее количество генов у человека (около 100 тыс.), суммарная мутабильность — величина немалая. Частота мутаций может значительно возрасти при действии на организм некоторых физических и химических факторов (мутагенов). Химические мутагены обнаружены среди промышленных ядов, инсектицидов, гербицидов, пищевых добавок и лекарств. Большинство канцерогенных веществ также обладают мутагенным действием. Кроме того, многие биологические факторы, например, вирусы и живые вакцины, а также гистамин и стероидные гормоны, вырабатываемые в организме человека, могут индуцировать мутации. Сильными мутагенами являются различные виды излучений (рентгеновские и γ-лучи, β-частицы, нейтроны и др.), способные вызывать генные и хромосомные мутации у человека, о чем свидетельствуют последствия аварии на ЧАЭС. К факторам, нарушающим постоянство генетической структуры популяций, относится и естественный отбор, вызывающий направленное изменение генофонда путем элиминации из популяции менее приспособленных особей или снижения их плодовитости. Рассмотрим влияние отбора на примере доминантной патологии — ахондроплазии (карликовости). Эта болезнь хорошо изучена в популяциях Дании. Больные имеют пониженную жизнеспособность и умирают в детском возрасте, т. е. устраняются естественным отбором из популяции. Выжившие карлики реже вступают в брак и имеют мало детей. Анализ показывает, что около 20% генов ахондроплазии не передается от родителей детям, а 80% этих генов элиминируются из популяции. Из этих данных следует, что ахондроплазия не оказывает существенного влияния на структуру популяции. Большинство мутантных генотипов имеют низкую селективную ценность и попадают под действие отбора. По данным В. Маккьюсика (1968), около 15% плодов погибают до рождения, 3% детей умирает, не достигнув половой зрелости, 20% умирают до вступления в брак, в 10% случаев брак остается бесплодным. Дата добавления: 2015-12-16 | Просмотры: 2438 | Нарушение авторских прав |