|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Дифференциальный диагноз геморрагических диатезовГеморрагические диатезы (ГД) представляют собой группу заболеваний наследственного или приобретенного характера, для которых характерны склонность к рецидивирующим кровотечениям и кровоизлияниям различной длительности и интенсивности.
К развитию геморрагического синдрома при ГД приводят нарушения различных звеньев сложного каскада гемостаза, наиболее часто — отсутствие или недостаток отдельных факторов свертывания крови (прокоагулянтов), избыток физиологических антикоагулянтов и фибринолитических агентов.
Система гемостаза обеспечивает предупреждение и остановку кровотечения путем поддержания структурной целости стенки кровеносных сосудов и достаточно быстрого их тромбирования при повреждениях. Эти функции обеспечивают 3 функционально-структурных компонента системы гемостаза: стенки кровеносных сосудов, форменные элементы крови, в первую очередь тромбоциты, и плазменные ферментные системы (свертывающая, фибринолитическая, калликреин-кининовая и др.).
Выделяют 2 механизма гемостаза:
1. Первичный (микроцитарный, сосудисто-тромбоцитарный) гемостаз, который обеспечивает остановку кровотечения из проксимальных и терминальных артериол, прекапилляров, истинных капилляров и венул посредством временного сосудистого спазма, адгезии и вязкого метаморфоза тромбоцитов с образованием тромбоцитарной пробки (белого тромбоцитарного тромба), ее последующего уплотнения и сокращения. Образовавшийся белый тромбоцитарный тромб стягивает поврежденные края мелких сосудов, удерживает их от дилатации и не пропускает жидкую часть крови.
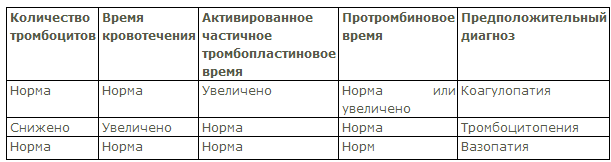
2. Вторичный (макроцитарный, окончательный) гемостаз, который обеспечивается системой свертывания крови и завершает полноценный гемостаз в макрососудах, начатый на сосудисто-тромбоцитарном этапе. Проводя дифференциальный диагноз ГД, следует учитывать данные анамнеза, физикального обследования больного и опираться на лабораторное выявление нарушений гемостаза, что позволяет верифицировать диагноз.
При опросе больного с геморрагическим синдромом необходимо:
1) установить приобретенный или наследственный характер заболевания;
2) уточнить сроки возникновения, давность, длительность и особенности течения заболевания (появление в раннем детском, юношеском возрасте или у взрослых людей, острое или постепенное развитие геморрагического синдрома, хроническое или рецидивирующее его течение);
3) выяснить причины возникновения или усиления кровотечения, локализацию, очередность появления элементов сыпи и изменения их окраски, эффективность проведенного лечения;
4) оценить наличие кровотечений после операций и травм, меноррагий, желудочно-кишечных и других кровотечений;
5) проанализировать наличие связи между появлением симптомов геморрагического синдрома и приемом лекарственных препаратов, прививками, различными патогенными воздействиями, сопутствующими заболеваниями (болезнями печени, инфекционно-септическим процессом, лейкозом, травмой, шоком и т. д.);
6) выяснить преимущественную локализацию, тяжесть и тип кровоточивости. При наличии информации о наследственном характере заболевания следует оценить выраженность симптомов у членов семьи (пенетрантность), наличие других генетических дефектов. Это связано с частым сочетанием наследственных ГД с другими аномалиями: телеангиэктазий — с гиперэластозом кожи, слабостью связочного аппарата, пролапсом митрального клапана; наследственной тромбоцитопении — с аномалиями скелета, нарушениями иммунной системы и пигментного обмена; гемофилии — с нарушениями цветного зрения.
Объективное обследование больного должно быть направлено на диагностику заболеваний, которые могли привести к ГД, а также оценку выраженности геморрагического синдрома. Необходимо учитывать, что клинические проявления ГД зависят от того, какое звено гемостаза поражено, а правильная оценка типа кровоточивости существенно облегчит дифференциальный диагноз при ГД, т. к. позволит целенаправленно применить тесты, верифицирующие диагноз.
При генетически обусловленной наследственной телеангиэктазии (болезни Рандю-Ослера) вследствие истончения базальной мембраны мелких кровеносных сосудов на слизистых оболочках, губах и коже образуются мелкие узловатые сосудистые образования, которые легко кровоточат и являются источником обильных и трудно купируемых кровотечений. Иногда телеангиэктазии сочетаются с мозжечковыми расстройствами и иммунной недостаточностью (синдром Луи-Бар).
При наличии видимых телеангиэктазий диагностика не сложна. Для выявления телеангиэктазий на слизистой оболочке пищеварительного тракта проводят эндоскопическое исследование. Показатели гемостаза обычно не отличаются от нормы.
Для приобретенных ГД сосудистого генеза (пурпуры Шенлейна-Геноха, гиперсенситивного васкулита, геморрагического васкулита инфекционно-токсического, инфекционно-воспалительного генеза и др.) характерен васкулитно-пурпурный тип кровоточивости, чаще с симметричным расположением высыпаний. Нередко выявляют другие виды сыпи (волдыри, папулы). Характерны также артралгия, гематурия, абдоминальные нарушения (болевой синдром, кишечные кровотечения), часто протекающие с лихорадкой. При легком течении нарушения гемостаза могут отсутствовать. При тяжелом течении, как правило, обнаруживают признаки хронического синдрома диссиминированного внутрисосудистого свертывания (ДВС-синдрома) — наличие фибрин-мономерных комплексов, положительные протаминсульфатный и этаноловый тесты. При фульминантной форме выявляют гипофибриногенемию, тромбоцитопению и коагулопатию потребления, что свидетельствует о наличии развернутого ДВС-синдрома.
Для ГД вследствие недостатка в крови тромбоцитов или их качественной неполноценности характерен петехиально-пятнистый тип кровоточивости с быстрым появлением геморрагий при надавливании на кожу, пальпации, сжатии руки манжетой тонометра (манжеточная проба), образование синяков вокруг мест инъекций, кровотечения из слизистых оболочек, меноррагии. Опасны кровоизлияния в головной мозг, о риске которых могут свидетельствовать геморрагии на коже лица и шеи. Возможны также кровоизлияния в сетчатку и яичники.
При обследовании больных с идиопатической тромбоцитопенической пурпурой (болезнью Верльгофа) выявляют значительное снижение уровня тромбоцитов в крови (менее 100•109 л) и гиперплазию мегакариоцитарного ростка в миелограмме. Характерны пойкилоцитоз тромбоцитов, укорочение времени их жизни, нарушение ретракции сгустка и увеличение времени кровотечения. Пробы на ломкость сосудов положительные.
Вторичные (симптоматические) тромбоцитопении развиваются при ряде заболеваний и состояний. В.М.Запорожан предлагает выделять:
1. Аутоиммунные тромбоцитопении при системных заболеваниях соединительной ткани и других заболеваниях иммунного генеза.
2. Гетероиммунные тромбоцитопении вследствие образования антител к поверхностному антигену тромбоцитов (медикаментозные иммунные тромбоцитопении и тромбоцитопении при вирусных заболеваниях).
3. Тромбоцитопенический синдром в результате гиперспленизма.
4. Тромбоцитопении вследствие влияния физических и химических факторов (ионизирующего облучения, электромагнитных волн, экзо- и эндогенных интоксикаций).
5. Тромбоцитопения при ДВС-синдроме.
6. Тромбоцитопении при заболеваниях системы крови (остром и хроническом лейкозе, гипопластической анемии, В12-дефицитной, иммунной гемолитической анемии).
Клинические проявления вторичных тромбоцитопений не отличаются от таковых при идиопатической тромбоцитопении, поэтому для исключения вторичного ее характера особо важное значение имеют данные анамнеза и исключение связи появления геморрагического синдрома с приемом лекарственных препаратов, профессиональными факторами, инфекционными и другими заболеваниями, которые могут сопровождаться тромбоцитопений. При вторичных тромбоцитопениях вследствие поражения мегакариоцитарного ростка помощь для уточнения диагноза могут оказать данные стернальной пункции (при лейкозе, гипопластической анемии).
Наличие функциональной неполноценности тромбоцитов необходимо заподозрить при наличии у больного типичных клинических признаков петехиально-пятнистого типа кровоточивости при нормальном количестве тромбоцитов в периферической крови. Тромбастения Гланцмана является наследственной тромбастенией, наследуемой по аутосомно-рецессивному типу, в основе которой лежит недостаток или дефицит гликопротеинового комплекса Iib-IIIa оболочки тромбоцитов, приводящий к нарушению связывания фибриногена и тромбоцитов. Болеют чаще женщины, заболевание проявляется в детстве. Характерны нарушения адгезии и агрегации тромбоцитов, ретракции кровяного сгустка и значительное увеличение длительности кровотечения при нормальном количестве тромбоцитов.
Среди наследственных ГД, связанных с нарушениями свертывающей системы крови, наиболее часто встречаются гемофилия А и В и болезнь Виллебранда, в связи с чем при наличии у больного гематомного типа кровоточивости диагностические мероприятия должны быть направлены в первую очередь на распознавание именно этих заболеваний. Все остальные наследственные нарушения свертываемости крови (дефицит V, VII, X и XI и других факторов свертывания крови) встречаются редко, а случаи дефицита и аномалий II, XII, XIII факторов свертывания крови, прекалликреина, высокомолекулярного кининогена — крайне редко, в связи с чем в данной статье не рассматриваются.
Гемофилии А и В обусловлены генетически детерминированным нарушением синтеза (реже аномалиями) VIII и IX факторов свертывания крови, гены которых локализуются в разных частях X-хромосомы и являются рецессивными. В связи с этим гемофилии А и В наследуются по типу, сцепленному с полом, и вызывают заболевание у лиц мужского пола, получивших от матерей патологически измененную Х-хромосому. По женской линии заболевание может передаваться в латентной форме на протяжении многих поколений, в связи с чем по данным анамнеза не всегда удается проследить наследование болезни. Кроме того, ген гемофилии А относится к часто мутирующим генам.
В большинстве случаев гемофилии А и В легко распознают в связи с наличием типичного гематомного типа кровоточивости. Повторяющиеся кровоизлияния в суставы и кости у лиц с этими заболеваниями могут приводить к развитию тяжелых деструктивных артрозов, контрактур, фиброзных анкилозов. Для больных гемофилией характерны обильные и длительные отсроченные (через 2–6 ч) посттравматические и послеоперационные, желудочно-кишечные, носовые и почечные (нередко с коликой и отхождением сгустков крови) кровотечения. Выраженность симптомов кровоточивости у больных с гемофилией А и В соответствует степени дефицита VIII и IX факторов свертывания крови. Если их содержание в крови ниже 1%, заболевание носит очень тяжелый характер, а при содержании более 5% — легкий.
Диагноз гемофилии базируется на результатах генетического анамнеза (наследование, сцепленное с мужским полом), данных клинической картины (гематомный тип кровоточивости) и лабораторных исследований (увеличение времени свертывания крови, признаки гипокоагуляции по данным аутокоагуляционного теста и увеличение активированного частичного тромбопластинового времени — АЧТВ). Дифференциальную диагностику гемофилий А и В осуществляют с помощью коррекционных тестов, в которых используется принцип разведения и коррекции нарушенного свертывания крови больного компонентами нормальной крови. Диагноз верифицирует количественное определение VIII и IX факторов свертывания крови.
Болезнь Виллебранда (ангиогемофилия) обусловлена наследуемым по аутосомному типу нарушением синтеза или аномалиями белкового кофактора VIII фактора свертывания крови (фактора Виллебранда). Болеют лица обоего пола, но у женщин заболевание протекает тяжелее. Дефицит фактора Виллебранда приводит к изменению не только коагуляционной активности VIII фактора свертывания крови, но и сосудисто-тромбоцитарного гемостаза (снижению адгезии тромбоцитов к субэндотелию и коллагену и их агрегации под влиянием ристомицина). Поэтому для пациентов с этим заболеванием характерен смешанный синячково-гематомный тип кровоточивости, а в крови, наряду с нарушением свертываемости, выявляют увеличение времени кровотечения, снижение адгезивности тромбоцитов и их ристомицин-агглютинации. Диагноз устанавливают на основании снижения содержания фактора Виллебранда в плазме крови и (или) в тромбоцитах. Дефицит К-витаминозависимых факторов свертывания крови (II, VII, IX и Х) может развиться при поражении печени у больных с циррозом, острым повреждением токсического и другого генеза (вследствие их недостаточного синтеза), при механической желтухе, тяжелых энтеропатиях и кишечном дисбактериозе (вследствие нарушения всасывания в кишечнике жирорастворимых витаминов, в том числе витамина К), геморрагической болезни новорожденных (вследствие временной депрессии выработки этих факторов в первые 4–7 суток после рождения), а также при избыточном приеме антикоагулянтов непрямого действия (вследствие конкуренции их с витамином К и вытеснения последнего из метаболизма К-витаминзависимых факторов свертывания крови с нарушением их карбоксилирования). Кровоточивость при ГД этой группы носит смешанный пятнисто-гематомный характер. Показатели лабораторных исследований свидетельствуют о выраженном снижении протромбинового индекса и значительном удлинении времени свертывания крови по данным АЧТВ при нормальных показателях тромбинового времени и содержании в крови фибриногена и тромбоцитов, при отрицательных тестах паракоагуляции (этанолового, протаминсульфатного). ГД вследствие передозировки прямых антикоагулянтов и фибринолитических препаратов могут явиться причиной кровоточивости смешанного типа (петехиально-пятнисто-гематомного), характеризующейся носовыми, почечными и желудочно-кишечными кровотечениями, а также высоким риском профузных желудочно-кишечных кровотечений у пациентов с пептической язвой, либо инсульта — у больных с артериальной гипертензией. Причины кровоточивости у больных, получающих препараты этих групп, ясны и обычно не требуют проведения дифференциального диагноза.
Таким образом, дифференциальный диагноз при наличии признаков нарушения гемостаза должен включать следующие этапы:
1. Опрос больного, который позволит выяснить наследственный или приобретенный характер заболевания, острое или хроническое его течение, выраженность нарушений гемостаза и провоцирующие факторы.
2. Физикальное обследование больного, позволяющее определить тип кровоточивости, который с высокой вероятностью указывает на поражение определенного звена гемостаза (сосудистого — при васкулитно-пурпурном типе, тромбоцитарного — при петехиально-пятнистом или коагуляционного — при гематомном и синячково-гематомном типах).
3. Применение лабораторных тестов, свидетельствующих о поражении различных звеньев гемостаза, в том числе целенаправленное количественное определение факторов свертывания в крови для проведения дифференциального диагноза внутри различных групп ГД.
Дата добавления: 2014-12-11 | Просмотры: 1511 | Нарушение авторских прав |