|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
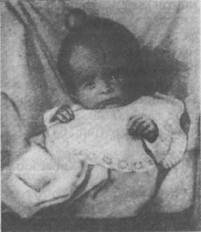
Аномалии развитияРассмотрим некоторые формы аномалий развития, связанные с влиянием вредных агентов на плод в процессе эмбриогенеза. Одной из тяжелых форм искаженного развития является так называемое уродство. Уродство представляет предмет изу- чения особой науки (тератологии) и в данном курсе может быть кратко охарактеризовано только с целью некоторого ознакомления. Причины уродства различны. Здесь может иметь значение наследственный фактор, но чаще это результат инфекций, интоксикаций, механических травм, действующих на разных стадиях эмбриогенеза. К уродствам могут быть отнесены случаи рождения детей без обеих рук или ног или с недоразвитием одной конечности. Наблюдаются случаи рождения близнецов со сросшимися частями туловища или скелета (сросшиеся близнецы). Тяжелым уродством являются такие формы, когда один из близнецов достиг нормального развития, а другой недоразвился в той или иной степени и прирос к телу первого в виде своеобразного нароста, имеющего иногда дифференцированное строение. Такие уроды могут иметь две головы, четыре ноги и т. д. Все эти лишние органы на теле одного из близнецов есть части тела недоразвившегося второго близнеца. Рассмотрим некоторые виды уродств, связанных с неправильным развитием нервной системы и системы желез внутренней секреции. Дефекты черепа, мозговая грыжа. В тяжелых случаях указанная аномалия заключается в резком недоразвитии черепных костей или их расщеплении, что приводит к уродливым формам развития черепа. Через образовавшиеся щели могут выпирать мозговая оболочка и мозговая ткань, образуя мозговую грыжу (рис. 62). Степень выраженности этих аномалий может быть различна. Неправильная форма черепа чаще не связана с какими-либо нарушениями нервной деятельности. Однако в отдельных случаях так называемый башенный череп, образующийся вследствие уменьшенного развития основания черепа, может сопровождаться ослаблением зрения в связи с атрофией зрительного нерва, сдавливаемого в костных отверстиях основания. Рис. 62. Мозговая грыжа
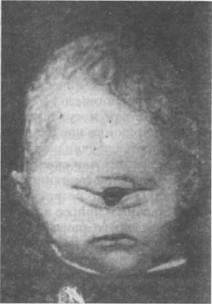
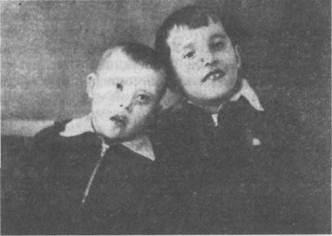
Рис. 63. Анэнцефалия Рис. 64. Циклопия Анэнцефалия.К этой группе тяжелых и редко встречающихся аномалий развития принадлежат случаи рождения плода, лишенного головы. Другие формы характеризуются отсутствием только больших полушарий или тем, что они находятся в зачаточном состоянии. Чаще подобные уроды родятся мертвыми или живут несколько часов или дней. Описанные в литературе отдельные случаи анэнцефалов, у которых наблюдались более продолжительные сроки жизни, от нескольких месяцев до 4 лет, характеризовались тяжелыми нарушениями различных органов (рис. 63). Циклопия- редко встречающийся дефект, характеризующийся тем, что у плода развивается только один глаз или одна глазная впадина, в которой заключены два глаза, сросшиеся вместе (рис. 64). Хромосомные нарушения Недоразвитие высших психических функций может быть обусловлено хромосомными аберрациями. Определенную часть детей с выраженной умственной отсталостью (олигофренией) составляют дети с болезнью Дауна, синдромами Шере-шевского—Тернера, Клайнфельтера и др. Болезнь Дауна. В основе заболевания в большинстве случаев лежит трисомия по 21-й паре хромосом (т.е. вместо двух имеются три хромосомы), в результате во всех клетках содержится 47 (вместо 46) хромосом. Основными проявлениями болезни являются умственная отсталость, врожденные пороки развития. Больные имеют своеобразный внешний облик (рис. 65): размеры черепа уменьшены, сглажены лобные и затылочные бугры; обращает на себя внимание раскосый разрез глаз, уплощенное переносье, широко расставленные глаза; рот маленький, всегда открыт, язык увеличен в размерах и испещрен бороздами; зубы мелкие, дистрофичные. Со стороны неврологического статуса отмечается недоразвитие моторных функций, особенно мелкой моторики, в связи с чем у детей задерживается психомоторное и речевое развитие. Дети начинают говорить поздно, при этом у них страдает фонетическая и лексико-грамматическая структура речи. По интеллектуальному развитию они значительно
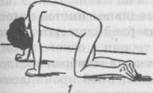
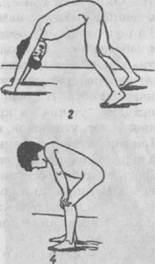
Рис. 65. Близнецы Слева — мальчик, страдающий болезнью Дауна; справа — нормальный ребенок (школьник) отстают от сверстников. В 5% случаев выявляется дебильность, 75% — имбецильность, в 20% — идиотия. С возрастом отмечается некоторое улучшение в общем состоянии ребенка. Часть детей воспитывается в дошкольных и школьных специальных учреждениях, другая часть — в домашних условиях, что обусловлено в ряде случаев желанием родителей. Исследования мозга детей с болезнью Дауна методом электроэнцефалографии показали снижение электрической активности мозга, вялость корковых процессов. Посмертное изучение мозга выявило уменьшение его веса, упрощение узора борозд и извилин, наличие свободных пространств между извилинами, недоразвитие лобных и теменных отделов коры. Характерны изменения в области клеточной структуры коры головного мозга: обнаруживается большое количество ганглионарных клеток и малое — пирамидных, значительное количество очагов демиелинизации. Синдром Клайнфельтера. В основе заболевания лежит по-лисомия по Х-хромосоме, в связи с чем общее количество хромосом достигает 47 или 48. Заболевание встречается только у лиц мужского пола и характеризуется умственной отсталостью и психической вялостью, а также нарушением полового развития (евнухоидизм, бесплодие). Степень снижения интеллекта может быть выражена в различной степени. Заболевание, помимо умственной отсталости, нередко сопровождается асоциальным поведением. Синдром Шерешевского—Тернера — форма первичной аге-назии или дисгенезия гонад. Заболевание у большинства больных обусловлено гоносомной моносомией 45-й Х-хромосомы (отсутствие одной из половых хромосом — X). Характеризуется разной степенью умственной отсталости, аномалиями соматического развития и низкорослостью. В последние десятилетия описаны различные заболевания в основе которых лежат хромосомные аномалии, так называемые делеции и транслокации. Все хромосомные аномалии проявляются нарушениями в строении костной и мышечной систем, внутренних органов и нервной системы. Синдром Штурге—Вебера — Краббе обусловлен трисомией по 22-й хромосоме. Заболевание сопровождается различной степенью психической отсталости. Возможны эпилептические припадки, сложные поражения нервной системы. Генетические расстройства Наряду с хромосомными нарушениями в настоящее время приобретают известность и генетические расстройства, обусловливаемые наследственными болезнями обмена веществ. Патология обмена веществ приводит к грубым морфологическим и функциональным изменениям в центральной и перифе- рической нервной системе. В зависимости от того, какой вид обмена веществ преимущественно нарушен, выделяют наследственные болезни обмена аминокислот, липидов, углеводов и т.п. Из группы наследственных нарушений обмена аминокислот наиболее изученной является фенилкетонурия. Биохимические изменения, наступающие вскоре после рождения ребенка, приводят к преимущественному поражению головного мозга и нарушению умственной деятельности. Фенилкетонурия. Заболевание описано в 1934 г. норвежским врачом А. Феллингом, наблюдавшим двух слабоумных братьев, в моче которых была обнаружена фенилпировино-градная кислота. Первоначально болезнь была названа «фе-нилпировиноградной олигофренией». Заболевание обусловлено генетически детерминированным дефицитом фермента фенилаланина-гидрококсидазы, что приводит к нарушению превращения фенилаланина в тирозин. Уже на 2—3-м месяце ждзни ребенка могут появиться признаки интоксикации (рвота, экзематозные изменения кожи), специфический запах мочи. Постепенно нарастает задержка психомоторного развития, появляются судороги, психические расстройства. У детей выражены парезы, гиперкинезы, задерживается развитие статических функций. К концу первого года жизни у детей выявляется глубокая умственная отсталость: они не узнают родителей, не играют с игрушками. М.Г. Блюмина выделила три варианта фенилкетонурии: 1) шизофреноподоб-ный; 2) простое слабоумие с преобладанием признаков общего психического недоразвити; 3) неврозоподобный вариант с явлениями повышенной заторможенности или же раздражительности. Судорожный синдром часто сопровождает данное заболевание, бывает особенно выражен у детей с глубокой умственной отсталостью. На ЭЭГ часто регистрируется низковольтная диз-метрия, недостаточная дифференциация зон мозга, низкая реактивность на афферентные раздражители. При патоморфоло-гическом исследовании выявлены низкая масса головного мозга, расширение желудочков и субарахноидального пространства. Микроскопически обнаружено недостаточное формирование или разрушение миелина, которое зависело от возраста больного и тяжести патологического процесса. Заболевание носит прогредиентный характер. Доктор А. Феллинг не только описал клинику фенилкетонурии, но и предложил специальную диагностическую реак- цию, благодаря которой можно выявить заболевание до проявления тяжелых признаков болезни. Им разработана лекарственная и диетотерапия, которая может обеспечить ребенку благополучное течение болезни. Дети, у которых рано выявили заболевание и начали лечение, находятся в специальном стационаре, а затем в специальной школе, что указывает на благополучный исход. Прогрессирующие мышечные дистрофии Данная группа заболеваний характеризуется прогрессирующей мышечной слабостью, атрофией мышц, двигательными нарушениями и имеет наследственную обусловленность. Миопатии — нервно-мышечные заболевания, характеризующиеся прогрессирующим развитием первичного дистрофического или вторичного (денервационного) атрофического процесса в скелетной мускулатуре, сопровождающимся мышечной слабостью и двигательными нарушениями. В основе наследственных миопатии лежат первичные обменные нарушения и расстройства микроциркуляции в мышечной ткани. Обычно при миопатии двигательные нарушения развиваются постепенно. Вначале у ребенка появляется слабость в верхних или нижних конечностях. В одних случаях патологический процесс может начинаться с мышц тазового пояса, в других — с плечевого. Формы с преобладанием поражения мускулатуры тазового пояса, а также спины и ног характерны для детей младших возрастов.В случае преобладания патологического процесса в мускулатуре плечевого пояса поражаются мышцы плеча и лопатки. Поражение мышц тазового пояса развивается в дошкольном или школьном возрасте. У детей рано появляется утомляемость при ходьбе, беге, возникают затруднения при подъеме по лестнице. Ребята часто падают. На первое место выступают расстройства движений и видимые атрофии, позже наряду с атрофиями мышц тазового пояса отмечается гипертрофия икроножных мышц (увеличение в объеме), которая зависит от отложения жира и разрастания соединительной ткани в них. Ноги в таких случаях приобретают форму бутылок. Очень характерно вставание больного из сидячего положения. Ребенок становится на четвереньки, опираясь о пол ладонями и стопами; затем отрывает ладони от пола и выпрямляется до вертикального положения (рис. 66). Особенно типична
Рис. 66. Способы вставания детей при миопатической атрофии походка миопатов со слабостью мышц та-.--g^^^g зового пояса и ног. Больные ходят, выставляя вперед живот, откидывая туловище назад и переваливаясь из стороны в сторону. Такую походку называют "утиной". Сила мышц в атрофированных конечностях резко снижена. Миопатия мускулатуры плечевого пояса чаще встречается в юношеском возрасте. Поражение мышц конечностей бывает симметричным. В результате атрофии лопаточных мышц лопатки выпирают назад и приподнимаются кверху. Плечи опущены вниз (рис. 67). Сила мышц резко понижена, в результате чего движения затрудняются, быстро наступает усталость. При ходьбе больные наклоняют корпус вперед, чтобы придать устойчивость голове. В этом случае мышцы предплечья и дельтовидные подвергаются гипертрофии. Характерно поражение лицевых мышц. Лицо своеобразно изменяется, губы становятся толстыми, веки не-
Рис. 67. Спинальная Дата добавления: 2015-01-18 | Просмотры: 1126 | Нарушение авторских прав |