|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Индуцированные радиацией доминантные мутации у мыши: мутации главных генов, не выявленные у человекаЭкспериментальная работа с млекопитающими, физиология развития которых ближе всего к человеку, показывает, как действие главного гена может быть скрыто за фенотипической изменчивостью организма. Такие главные гены идентифицируются с помощью подходящих экспериментальных скрещиваний или на основе феногенетического анализа индуцированных мутаций. Мы обсудим один пример, который важен также для оценки риска индуцированных мутаций у человека [865; 640]. Генетические дефекты, связанные с доминантными мутациями, можно выявить путем сравнения потомков первого поколения от опытных и контрольных животных. Однако для многих признаков трудно провести различие между вновь возникшими мутациями и внутрилинейной изменчивостью. Эта трудность была преодолена для некоторых скелетных аномалий у мыши. В мутационном эксперименте аномалии, наблюдавшиеся в поколении F1, можно разделить на те, которые проявляются крайне редко на протяжении всего эксперимента (класс 1), и на такие, которые встречаются много чаще (класс 2). Разумная рабочая гипотеза (при исследовании многих сотен особей) заключается в предположении, что большинство очень редких аномалий (класс 1) имеют мутационное происхождение, тогда как большинство частых аномалий (класс 2) обусловлены внутрилинейной изменчивостью. Согласно этой гипотезе, мутагенные факторы типа ионизирующей радиации должны повысить количество первично очень редких (класс 1) аномалий. Это подтверждено в достаточном количестве экспериментов с ионизирующей радиацией и химическими мутагенами. Фенотипически большинство редких (класс 1) и более распространенных (класс 2) аномалий представляют собой множественные минорные варианты скелета. Некоторые из них, например аномалии позвоночника, вредны в разной степени. Относительно 31 аномального варианта скелета с помощью экспериментальных скрещиваний было подтверждено, что они вызываются доминантными мутациями. Выделим две характеристики этих доминантных мутаций. 1. Некоторые или все аномалии мутационного происхождения имеют низкую пенетрантность. 2. Морфологически можно выявить лишь небольшую долю этих мутаций, и те, которые можно выявить, не проявляются у большинства носителей гена. Экспериментальные скрещивания показали, что в потомстве особей F1 вероятность выщепления аномальных фенотипов оказывается гораздо ниже ожидаемой 0,5. Сопоставляя эти результаты с данными 3. Формальная генетика человека 257
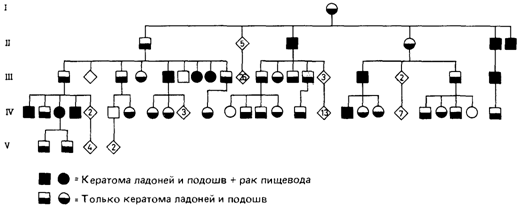
по доминантным мутациям у человека, можно предположить, что трудности выявления таких мутаций у мыши не столь актуальны для человека, тщательное медицинское обследование позволяет идентифицировать большинство этих мутаций. При рассмотрении доминантных генов с неполной пенетрантностью очень важной проблемой остается установление низкой пенетрантности аутосомных мутаций. Кроме того, часто регистрируемые аномалии обнаруживают поразительную степень изменчивости между особями, несущими мутантные гены, идентичные по происхождению. С другой стороны, фенотипические спектры одной и той же аномалии у носителей разных мутаций сильно перекрываются. Фенотипические проявления некоторых мутаций были почти идентичными. Для сравнения этих результатов с генетическими данными у человека необходимо было бы выявить у него уродства скелета, сходные с таковыми у мыши. Попытки такого рода уже предпринимались, но из-за неразработанности вопросов генетики скелетных аномалий человека большого успеха достичь не удалось. Недавний «всплеск» новых исследований в этой области [774], возможно, приведет к пересмотру накопленных данных и некоторых обобщений. Обескураживает, однако, то обстоятельство, что до сих пор не удается найти у мыши скелетные мутации, идентичные мутациям у человека. Описанные эксперименты оставили нерешенными некоторые вопросы, в частности вопрос о возможности незначительных хромосомных перестроек. В порядке рабочей гипотезы можно выдвинуть следующее предположение: у человека существует большое количество доминантных мутаций, вызывающих широкий диапазон морфологической изменчивости и, вероятно, оказывающих влияние на здоровье. Однако современные методы феногенетического анализа весьма несовершенны и не дают возможность раскрыть генетическую основу этой изменчивости. 3.6.2.6. Идентификация элементарных клинико-генетических вариантов моногенного наследования с использованием дополнительных фенотипических критериев Иногда в пределах большой гетерогенной группы больных можно выделить отдельные формы патологии с отчетливо менделевским наследованием. Это удается сделать на основе детального клинического изучения, лабораторных исследований и генетического анализа. Данные, полученные при этом, позволяют отделить генетические случаи от негенетических. Подобные результаты были получены для умственной отсталости [2157], глухоты [669] и слепоты [670]. С развитием и совершенствованием нозологии в области психоневрологии и с повышением уровня клинических исследований некоторые задержки умственного развития, которые ранее относили к общей группе клинически недифференцированных форм, теперь можно достаточно четко классифицировать. В качестве примера весьма распространенного признака можно упомянуть Х-сцепленную форму умственной отсталости с маркерной «ломкой» Xхромосомой [2220]. Успешными в этом смысле были также исследования слепых и глухих детей, живущих при лечебных учреждениях. Оказалось, что около 50% всех случаев глухоты и слепоты имели генетическую природу. И практически все эти случаи были скорее менделевскими, чем мультифакториальными. Среди них было найдено много разных клинических форм с простым типом наследования. Почти всегда в рамках мультифакториального заболевания можно идентифицировать редкие менделевские варианты. Так, Х-сцепленная недостаточность фермента HGPRT составляет 1% всех случаев подагры. Некоторые случаи гипертонической болезни вызываются редкой наследственной феохромоцитомой. Язвенная болезнь желудка и двенадцатиперстной кишки выступает как часть симптомокомплекса при болезни Золлингера-Эллисона. Рак пищевода иногда возникает при генетически обусловленных кератомах одновременно на ладонях и подошвах (рис. 3.66). 258 3. Формальная генетика человека
Имеется ряд синдромов, при которых рак оказывается частью более сложной плейотропной картины (разд. 5.1.6). Иногда в семьях наблюдается доминантное наследование более или менее распространенных форм рака. В этом случае раннее начало и множественные поражения помогают отделить эти проявления главного гена от обычных типов рака. В родословной на рис. 3.66 возраст проявления рака приходился на 34, 37, 38, 43, 44, 45, 46, 52 и 63 года. Однако все эти случаи, кроме последнего, очень необычны для рака пищевода. В дерматологии наблюдается множество как изолированных, так и семейных случаев доброкачественных и злокачественных опухолей. Установлено правило [84], согласно которому единичные опухоли у одного больного имеют негенетическое происхождение, тогда как множественные опухоли имеют тенденцию наследоваться, причем часто обнаруживают аутосомно-доминантный тип наследования (см. также раздел 5.1.6). Дата добавления: 2015-12-16 | Просмотры: 815 | Нарушение авторских прав |