|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Жовте насіння зелене насіння
Рисунок 1.1 - Схема, яка ілюструє другий закон Г.Менделя (1АА:2Аа:1аа)
Появу зеленого насіння у другому поколінні Г.Мендель пояснив тим, що успадковуються не самі ознаки, а фактори (гени). На основі цього був сформульований закон чистоти гамет: Алельні гени, знаходячись у гетерозиготному стані, не зливаються, не змінюють один одного і, не втрачаючи своєї індивідуальності, передаються в гамети. Третій закон Менделя - закон незалежного комбінування ознак: при схрещуванні гомозиготних особин, які відрізняються двома (і більше) парами альтернативних ознак, у другому поколінні спостерігається незалежне успадкування і комбінування ознак, якщо гени цих ознак розташовані в різних парах хромосом. Схема (рис. 1.2), яка ілюструє третій закон Г.Менделя на насінні гороху (решітка Пеннета, рис. 1.3): А – домінантний алель жовтого кольору насіння; а – рецесивна алель зеленого кольору насіння; В –домінантна алель гладенької форми насіння; в – рецесивна алель зморшкуватої форми насіння. Гомозигота Гомозигота Жовта гладенькаЗелена зморшкувата Р ААВВ х аавв Гамети АВ ав F1 АаВв Гетерозиготи жовті і гладенькі Р АаВв х АаВв
Рисунок 1.2 -Схема, що ілюструє третій закон Менделя
Цитологічною основою цього закону є мейоз. Не гомологічні хромосоми у мейозі розходяться і можуть комбінуватися в будь-яких поєднаннях. Таблиця 1.1. - Решітка Пеннета до третього закону Менделя
Установлені Г. Менделем закономірності спадкування ознак, відповідно до сучасної термінології, можна зформулювати таким чином: 1. Кожна ознака в організмі контролюється парою алелей певного гена. 2. При мейозі кожна пара алелей розщеплюється і кожна гамета одержує по одному алелю з кожної пари. 3. При утворенні чоловічих і жіночих гамет у кожну з них може потрапити будь-який алель з однієї пари разом із будь-яким алелем з іншої пари. 4. Кожний алель передається з покоління в покоління як дискретна незмінна одиниця спадковості. 5. Материнський і батьківський організми так само беруть участь у передаванні своїх спадкових факторів нащадкам. 6. Нове покоління одержує не готові ознаки, а тільки спадкові фактори – по одному алелю (для кожної ознаки) від кожної батьківської особини (Кулікова Н.А., Ковальчук Л.Є., 2004). Генетична інформація закладена в послідовності основ ДНК, і ця послідовність кодує послідовність амінокислот у білку. Нуклеїнові кислоти складаються з хімічно зв’язаних нуклеотидів. Нуклеотид складається з трьох компонентів: азотистої основи, пентоди та залишку фосфорної кислоти (фосфатної групи). У нуклеїнових кислот виділено два типи пентод (ДНК та РНК). Азотисті основи діляться на піримідинові (цитозин (С), тимін (Т)) та пурини (аденін (А), гуанін (G)). У РНК замість тиміну – урацил. Нині загальновизнаною є модель ДНК у вигляді подвійної спіралі, запропонована Джеймсом Уотсоном та Френсісом Кріком (1953). У кожному ланцюгу основи спрямовані всередину спіралі так, що пурин завжди розташований проти піримідину. Гуанін утворює водневий зв’язок тільки з цитозином, аденін – тільки з тіаміном. Такі реакції називаються компліментарністю (спарюванням). Встановлено, що у будь-якій молекулі ДНК сума нуклеотидів, що містять пурини, дорівнює сумі нуклеотидів, що містять піримідини (Чаргафф Е.). Структура подвійної спіралі ДНК руйнується при нагріванні (денатурація або плавлення). За певних умов при цьому можливе відновлення (ренатурація). Поряд з ДНК у клітині наявні молекули РНК. Основна їх роль – трансляція генетичної інформації з утворенням білків. У клітинах наявні такі види РНК: рибосомальна (рРНК), матрична або інформаційна (мРНК або іРНК), транспортна (тРНК), затравна РНК праймосома). Однією із особливостей генетичного матеріалу є здатність до точного самовідтворення. Реплікація – процес самовідтворення ДНК. Генетичний код – система запису спадкової інформації в молекулах нуклеїнових кислот шляхом чергування послідовностей нуклеотидів. Одиниця генетичної інформації, що визначає, яка з амінокислот буде вбудовуватися в молекулу білка, який синтезується, називається “кодон”. Встановлена триплетність коду, визначені загальні властивості коду. Перенесення генетичної інформації здійснюється від ДНК до ДНК (шляхом реплікації), від ДНК через мРНК до білка. Крім того, можливе перенесення генетичної інформації від РНК до ДНК (зворотна транскрипція). Процес зчитування генетичної інформації названий транскрипцією (переписуванням) здійснюється ферментами РНК-полімеразами. Процес, внаслідок якого виникають нові комбінації послідовностей ДНК, проходить перерозподіл генів, називається рекомбінацією (поєднанням). Процес обміну ділянками між гомологічними (схожими) хромосомами називається гомологічною рекомбінацією, або кросинговером, тобто схрещуванням. Негомологічні рекомбінації – здатність певних елементів переміщатися в геномі.
Установлено, що несприятливі фактори призводять до появи вставок, делецій, розривів, блокування реплікації і загибелі клітин. Однак ДНК здатна усувати (репарувати) ушкодження, які виникли в стуктурах. Під впливом різних факторів проходять зміни послідовності ДНК, які називають мутаціями. Найбільш ушкоджувальну дію мають мутації, які спричинюють до утворення неінформативного білка, заміни нуклеотидів, за яких утворюються термінальні (стоп-кодони). Відомі також мутації, які не приводять до помітних порушень функції генів. Успадкування груп крові в людини відбувається за типом множинних алелей. Локус автономного гена позначається ІА, ІВ, І0 (або і). Алелі ІА, ІВ контролюють синтез відповідно антигена А і антигена В, а І0 – ніякого. Антигени містяться на поверхні еритроцитів та лейкоцитів, тромбоцитів, клітин тканин. Залежно від їх комбінацій існують 4 групи крові (4 фенотипи). Відмінність між ними - в наявності чи відсутності аглютиногенів А, В та аглютинінів a (альфа) і b (бета) у плазмі крові. Чотирьом фенотипам відповідають шість генотипів (І(0)-іі, ІІ (А) – ІАІА, ІАі, ІІІ (В) –ІВІВ, ІВі, ІY (АВ)- ІАІВ). Оскільки групи крові генетично обумовлені й не змінюються протягом життя, то за групою крові (рис. 3) можна встановити, чи виключити батьківство в цьому випадку (за групою крові неможливо стверджувати, що саме цей чоловік є батьком дитини).
Таблиця 1.2 - Можливі групи крові батька при відомій групі крові дитини та матері
Необхідно пам’ятати, що в осіб з ІY (АВ) групою крові в 0,1-0,2% випадків спостерігається особливе положення генів – цис-положення, коли обидва гени ІА та ІВ знаходяться в одній хромосомі. Тоді у шлюбі такої людини з особою, що має І (0) групу крові, можливе народження дітей з І (0) групою крові, що необхідно враховувати при проведенні судово-медичної експертизи. Такі ознаки, як група крові АВ0, резус – фактор, праворукість, мають у людини пенетрантність 100%. Частота груп крові у популяціях людей різних частин світу відрізняється. Так, у росіян (Моcква) група крові 0(І) спостерігається в 33,3%, А(ІІ) –в 37,4%, В(ІІІ) – в 22,8%, АВ(ІV)- в 6,5%. В аборигенів Австралії не зустрічаються групи В(ІІІ), АВ(ІV). У популяції Бороло (Бразилія) та Майя (Південна Америка) зустрічається тільки група 0(І). Основні постулати медичної генетики: 1. Не існує межі між спадковою мінливістю, що приводить до варіацій спадкових ознак, і мінливістю, в результаті якої виникають спадкові захворювання. 2. У процесі тривалої еволюції людство накопичило величезний “вантаж” різноманітних мутацій. 3. На ступінь прояву спадкових хвороб впливають як генотип, так і навколишнє середовище. 4. Спадкова обтяженість включає шкідливі мутації. Не існує різкої межі між спадковою мінливістю, що призводить до нормальних варіацій ознак, та спадковою мінливістю, що приводить до спадкових хвороб. Хвороби виникають у випадках, коли мутації порушують життєво важливі функції організму. Одна із важливих концепцій медичної генетики - концепція еволюційного накопичення патологічних мутацій у популяції людини (Homo sapiens). Ефектами генетичного вантажу в людини є: - Балансований поліморфізм (у популяції представлені дві форми алелей одного гена; частота рідкого алеля становить не менше 1 %). - Летальність - загибель гамет, зигот, ембріонів, плодів, смерть дітей. - Знижена фертильність - знижене відтворення потомства у сім'ях зі спадковою патологією. Мутації з клінічно подібними проявами до хвороб людини трапляються і в тваринному світі (гемофілія, м’язова дистрофія тощо). Спадкові хвороби у людей спостерігалися і тисячоліття тому, про що свідчать археологічні розкопки. Мутаційна мінливість є основою розмаїття ознак. Коли ми говоримо про наявність мутації, то розуміємо, що наявні порушення, які негативно впливають на дану особу. Для більшості осіб зі спадковою патологією характерні зниження фертильності за рахунок порушення репродуктивної функції та зниження кількості дітей у таких сім’ях. Наслідком мутацій є соціальна дезадаптація хворих (інвалідність), підвищена потреба в медичній допомозі та зниження тривалості життя. З генетичної точки зору, у людини можна виділити: спадкові хвороби, хвороби зі спадковою схильністю (мультифакторіальні) та неспадкові хвороби. Спадкові хвороби - це хвороби, що виникають унаслідок пошкодження генетичної інформації. Мультифакторіальні хвороби - це хвороби, які розвиваються в осіб з певною генетичною характеристикою під впливом факторів довкілля. Причинами спадкових хвороб є геномні, хромосомні та генні мутації. Геномні мутації пов'язані зі зміною кількості хромосом. Хромосомні мутації пов'язані зі зміною структури хромосом. Генні мутації - це молекулярні зміни на рівні ДНК. Для спадкових хвороб характерним є клінічний поліморфізм, при якому спостерігається різноманітність клінічних і лабораторних проявів хвороби. Причинами клінічного поліморфізму є генетичні фактори і фактори навколишнього середовища. Генетичні причини клінічного поліморфізму зумовлені генетичною гетерогенністю чи модифікуючим впливом генетичної конституції (взаємодією генів). Виділяють вроджені хвороби. Природжені хвороби - це спадкові й неспадкові хвороби, що спостерігаються при народженні дитини. Не всі спадкові хвороби є природженими, оскільки приблизно половина їх виявляється пізніше - у дитячому, юнацькому, зрілому чи літньому віці. В основу генетичної класифікації спадкових хвороб покладено етіологічний принцип. Виділяють такі групи спадкових хвороб: - генні; -хромосомні; -мультифакторіальні; -хвороби соматичних клітин; -хвороби генетичної несумісності матері та плода. Малі аномалії розвитку (МАР) становлять близько половини усіх морфологічних ознак, які використовуються для диференціальної діагностики синдромів множинних вад розвитку (МВР). До МАР належать морфологічні зміни органа, які виходять за варіанти норми чи знаходяться близько між нормою та патологією. Виділяють малі природжені вади, малі варіанти норми (сімейні чи малі аномалії) і порушення розвитку, що минають. Малі природжені вади - анатомічні дефекти, що зазвичай не потребують хірургічної корекції, але іноді є косметичним недоліком. Малі варіанти норми (сімейні, або малі, аномалії) — варіанти фенотипу, відсутні у синдромі мальформації (вади розвитку); часто трапляються у родині, етнічній групі, до якої належить індивід. За клінічним ступенем інформативності для діагностики МАР поділяють на неспецифічні, відносно специфічні, високоспецифічні й ознаки, не характерні для даного синдрому. Значна частина МАР є неспецифічними. Хромосомна теорія спадковості обґрунтована і сформульована американським генетиком Т.Морганом та його школою в 1910-1925 роках. За розроблення хромосомної теорії Т.Морган одержав Нобелівську премію (1933). Це основна теорія генетики, за якою матеріальними носіями спадковості є хромосоми, в яких лінійно розташовані гени.
Основні положення хромосомної теорії:
Хромосомний комплекс людини складається із 23 пар хромосом, з яких 22 пари – називаються аутосоми, а 23-тя пара – статеві хромосоми. Хромосома складається із ДНК та білка, зв’язаних разом. ДНК щільно скручена на білковому стрижні (розміщена в ядрі діаметром 0,005 мм, але якщо повністю розгорнути, то довжина становитиме майже 2 метри. Хромосоми – це нуклеопротеїдні структури. Вони подвоюються перед кожним клітинним поділом, а потім порівну розподіляються між дочірніми клітинами. В усіх клітинах особини одна і та сама хромосома має ту саму форму і несе однакову інформацію. До складу хромосом входять ДНК (40%), гістонові білки (4%), негістонові білки (20%) і незначна кількість РНК. Жіноча стать представлена двома Х-хромосомами, а чоловіча ХY-хромосомами. Кожна хромосома має індивідуальну форму та індивідуальний генетичний зміст. На Міжнародному симпозіумі у Денвері (США) на пропозицію проф. Патау 23 пари хромосом людини були розподілені на 7 груп залежно від розміру (рис. 1.5): А(1, 2, 3), В(4, 5), С(6, 7, 8, 9, 10, 11, 12), D(13, 14, 15), E(16, 17, 18), F(19, 20), G(21, 22). Окремо виділені Х- та Y-хромосоми. Патау було запропоновано розрізняти хромосоми не тільки за величиною, але і за формою залежно від місця розташування центромери (метацентричні, субметацентричні, акроцентричні). Метацентрична хромосома ділиться центромерою на два практично рівних плеча, субметацентрична –це коли центромера зміщена до одного з полюсів, а при акроцентричній хромосома має одне дуже коротке плече (центромера розташована близько до полюса). Зберігаючи генетичну інформацію, хромосоми змінюють свою довжину під час проходження клітинного циклу. У кожній хромосомі наявна велика кількість генів, розташованих у хромосомах лінійно, причому у кожного гена наявне чітко визначене місце (займають до 25% усієї довжини ДНК). Гени є носіями спадкових ознак. Один ген формує одну ознаку, проте часто ген впливає на формування кількох ознак. Існують також ознаки, які формуються дією кількох генів. Процес трансформації генетичної інформації в білки здійснюється шляхом копій послідовностей нуклеотидів ДНК у послідовність ланцюга РНК, яка називається матричною, або інформаційною РНК (мРНК). Процес транскрипції виконується ферментами, які названі РНК-полімеразами. Ген – функціональна одиниця ДНК, яка містить інформацію про синтез одного поліпептидного ланцюга або РНК (рибосомної, транспортної). Сам термін „ген” з’явився в 1909 році і запропонований датським біологом Вільгельмом Йогансоном. Кожний ген містить близько 1000 пар основ, і їх послідовність у кожному гені унікальна.
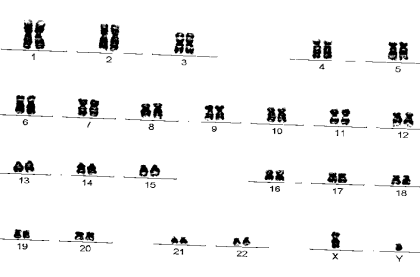
Рисунок 1.3 - Нормальний чоловічий каріотип (G-фарбування) Таблиця 1.3 - Класифікація хромосом людини
Уся сукупність генетичної інформації кожного клітинного ядра називається геномом. Сукупність генів, які визначають спадкову конституцію, називаються генотипом. Реальні властивості особи (сукупність усіх індивідуальних особливостей організму) - фенотип - виникають унаслідок взаємодії генотипу та впливу навколишнього середовища. Компонентами стабільності геному є: 1. Дублювання структурних елементів геному. 2. Матричний принцип біосинтезу ДНК (реплікація) й РНК (транскрипція). 3. Здатність до репарації - відновлення ушкоджених генетичних структур. 4. Генетичний контроль генної активності. Загальні властивості генетичного коду: -універсальність (ідентичний для всіх живих організмів); -триплетний (кожна амінокислота кодується трьома нуклеотидами); -не перекривається (кожний кодон складається з трьох нуклеотидів і наступний, представлений такими трьома нуклеотидами); -не містить будь-яких розділових знаків між триплетами (без ком); -колінеарний (порядок розташування кодонів у мРНК збігається з порядком кодованих ними амінокислот у білку, що синтезується); -має фіксовану стартову точку (зчитування починається на одному кінці, а закінчується на іншому); -вироджений (усі амінокислоти, за винятком метіоніну та триптофану, мають більше ніж один кодон); -із трьох нуклеотидів кодону переважне значення мають два перших, третій може варіювати; -у середньому кожна амінокислота кодується трьома триплетами. Перенесення генетичної інформації здійснюється: -від ДНК до ДНК шляхом реплікації, -від ДНК через мРНК до білка, -від РНК до ДНК за допомогою процесу зворотної транскрипції. Несприятливі чинники можуть призводити до структурних змін ДНК і навіть загибелі клітин. Мутації спричиняють значні різноманітні прояви спадкової патології, а в деяких випадках можлива внутрішньоутробна смерть ембріона чи плода. Однак ДНК здатна репарувати (відновлювати) ушкодження у своїй структурі. Виділяють 4 механізми репарації (відновлення) ДНК: 1. Фотоактивація. В деяких клітинах, наприклад кишкової палички, є фермент, який активується світлом та виправляє пошкодження в ДНК. 2. Ексцизія - чотириступеневий процес відновлення (одна нитка ДНК розрізується ферментом (УФ-ендонуклеазою), а фермент ДНК-полімераза видаляє нуклеотиди біля розрізу, включаючи пошкоджені ділянки. 3. ДНК-полімераза замінює видалені нуклеотиди правильми, використовуючи нерозрізану комплементарну нитку як матрицю. 4. SOS-репарація. Дефекти в ДНК інколи можуть бути замінені без урахування компліментарності основ. Це здійснюється при масивних пошкодженнях у ДНК і дозволяє клітині відновити пошкодження та вижити, але збільшує ризик утворення мутацій. Мутації розділяють на три класи: 1) мутації, що призводять до повної втрати функції (loss-of-function); 2) мутації, що супроводжуються кількісними змінами відповідних мРНК і первинних білкових продуктів; 3) домінантно - негативні мутації, що змінюють властивості білкових субодиниць, які мають пошкоджувальну дію на життєздатність або функціонування експресивних типів клітин (gain-of- function). Є багато причин мутацій. Деякі виникають спонтанно, в результаті помилок при реплікації та репарації ДНК. Інші індукуються, виникають в результаті дії мутагенів та факторів навколишнього середовища. Існують три типи мутагенів: -іонізуюча радіація, яка порушує нормальну послідовність основ ДНК, вибиваючи із них пари основ; -неіонізуюча радіація (ультрафіолетове опромінення, яке приводить до зшивання двох розташованих поряд вимінів у нитки ДНК, що блокує реплікацію ДНК та потребує її репарації); -хімічні мутагени (змінюють пари основ). Формою спадкової мінливості є однонуклеотидний поліморфізм. Саме його існування підтверджує відсутність різниці між спадковою мінливістю, що викликає нормальні варіації, і спадковою мінливістю, яка спричиняє спадкову патологію. Клонування і секвенування геному допомогло встановити, що не всі зміни нуклеотидної послідовності гена спричиняють патологічні ефекти. При змінах у хромосомах порушується синтез білка, ферментів, гормонів і т.д. Існують генні, або точкові мутації, які лежать в основі спадкових хвороб обміну речовин (ензимопатії). Існують також хромосомні й геномні мутації, які характеризуються змінами в хромосомах (структурі або кількості хромосом). Вони становлять матеріальну основу хромосомних хвороб. Пренатальний період розвитку людини поділяють на три головні стадії: преембріональну, ембріональну і плідну (фетальну). Виділяють критичний період розвитку - це період, протягом якого зародок людини найбільш чутливий до впливу тератогенних факторів: наприкінці 1-го - на початку 2-го тижнів гестації і між 3-м та 6-м тижнями ембріогенезу. Термінаційний тератогенний період - граничний термін в ембріональному формуванні органа, протягом якого пошкоджувальний фактор може викликати в ньому розвиток вади. Залежно від об’єкта та терміну впливу вражаючих факторів виділяють: -гаметопатії – результат ураження статевих клітин, що призводить до порушення спадкових структур. До них відносять всі спадково обумовлені вроджені вади, в основі яких лежать мутації в статевих клітинах батьків хворого. Бластопатії - результат ураження бластоцитів, тобто зародка людини в період перших 15 днів після запліднення (до завершення диференціювання зародкових листків). Результатом бластопатій є двійникові вади (близнюки, що зрослися), циклопія (наявність одного чи двох очних яблук, в одній орбіті, що розташована по середній лінії обличчя, частина мозаїчних форм хромосомних хвороб – також результат бластопатій; -ембріопатії – результат впливу тератогенного чинника на ембріон у період з 16 дня до 8-9 тижнів вагітності. До цієї групи відносять діабетичні, алкогольні, медикаментозні ембріопатії, а також вади, зумовлені вірусом краснухи. -фетопатії- результат ураження плода в період з 9 тижнів до періоду народження. Вади в цей період порівняно нечасто (крипторхізм, відкритий Боталовий проток, пренатальна гіпоплазія якогось органа чи всього плода). У період органогенезу наявні проміжки, протягом яких вплив тератогенних факторів може викликати порушення правильного формування органів. Чутливість закладок різних органів до дії тератогенних факторів різна. Найбільш рано формуються вади ЦНС (2-6 тижнів) та серця (3-6 тижнів). Практично всі вади розвитку формуються в період до 7-9 тижнів внутрішньоутробного розвитку. Вроджені вади розвитку, які виникли після закінчення основного періоду органогенезу, проявляються головним чином у вигляді зупинки в розвитку структур органа – гіпоплазії, затримкою переміщення органа (крипторхізм), вторинними змінами (деформації кінцівок при маловодді). На Празькій конференції (1971) з цитогенетики була узгоджена спеціальна номенклатура опису каріотипу людини. Передусім вказують загальне число хромосом (46, 45, 47 і т.п.), потім набір статевих хромосом (ХХ, ХУ). Після цього записують групу або номер зайвої чи відсутньої хромосоми (+21, -Х), далі вказують структурні зміни в тих чи інших хромосомах (делеція – del, довільна хромосома - der, дуплікація - dup, інcерція (вставка) - ins, інверсія - inv, реципрокна трансформація - rep, робертсонівська транслокація (центричне злиття) - rob, рекомбінантна хромосома - rec, тандемна транслокація - tan, термінальний (кінцевий) - ter, кінець короткого плеча - pter, кінець довгого плеча - qter, розрив без сполучення -:, розрив і з’єднання -::, від... до ---, ізохромосома - i, кільцева хромосома - r, транслокація - t. Приклад: 46,ХХ –каріотип нормальної жінки, 46,ХУ – каріотип нормального чоловіка, 47, ХХУ –каріотип чоловіка із синдромом Клайнфельтера, 47,ХХ,+21- жінка з каріотипом, що включає до себе додаткову 21-шу хромосому, 45, Х0 – каріотип жінки із синдромом Шерешевського-Тернера. 46,Х,і(xq) – каріотип жінки, що має 46 хромосом, одна Х-хромосома пошкоджена і одна ізохромосома зa довгим (q) плечем. Ознаки, успадкування яких підлягає закономірностям, що встановлені Г.Менделем, називаються менделювальними. Всі мендилювальні ознаки дискретні й контролюються одним геном (моногенні). Домінантні менделювальні нормальні ознаки людини: -карі очі, -великі очі, -темне волосся, -колір шкіри смуглий, -ластовиння є, -косий розріз очей, -ніс із горбинкою, -широка щілина між різцями, -зуби великі, виступають уперед, -ямочки на щоках, -лисина у чоловіків, -біле пасмо волосся, -мочка вуха вільна, -повні губи, -краще володіння правою рукою, -кров резус-позитивна, -антигени системи АВ0, -здатність згортати язик трубочкою. Рецесивні нормальні ознаки успадкування: -блакитні очі, -маленькі очі, -світле волосся, - колір шкіри білий, -прямий розріз очей, -прямий ніс, -вузька щілина між різцями або її відсутність, -звичайна форма і розміщення зубів, -відсутність ямочок на щоках, -рівномірна пігментація волосся, -відсутність ластовиння, -мочка вуха приросла, -губи тонкі, -краще володіння лівою рукою, -кров резус – негативна, -група крові за системою АВ0 – 0(1) -нездатність згортати язик трубочкою. Патологічні ознаки, які успадковуються домінантно: -карликова хондродистрофія, -полідактилія, брахідактилія, -поліпоз товстої кишки, -еліптоцитоз (еліпсоподібна форма еритроцитів). Менделювальні патологічні ознаки, які успадковуються рецесивно: -гемофілія, -дальтонізм (кольорова сліпота), -альбінізм (відсутність пігментів), -фенілкетонурія, -галактоземія, -фруктозурія. Усі менделювальні ознаки контролюються одним геном.
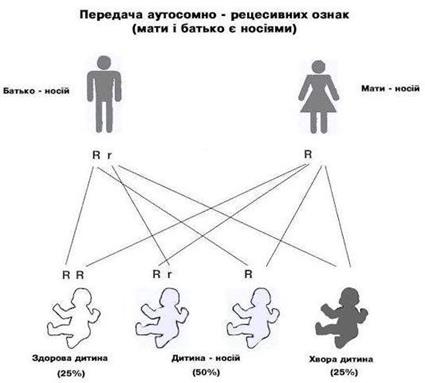
Ознаками автосомно-домінантьного типу успадкування є: -хвороби трапляються у кожному поколінні (вертикальний тип спадковості); -співвідношення хворих і здорових 1:1, здорові діти хворих батьків народжують здорових дітей; -співвідношення хворих хлопчиків і дівчаток 1:1, хворі чоловіки і жінки однаково передають хворобу своїм дітям - як хлопчикам, так і дівчаткам; -чим тяжча хвороба позначається на репродукції, тим більша пропорція спорадичних випадків (нових мутацій); - гомозиготи народжуються від двох хворих батьків, перебіг хвороби у них важчий, ніж у гетерозигот. Найбільш поширені хвороби, які успадковуються за автосомно-домінантним типом: нейрофіброматоз 1-го та 2-го типів, синдроми Марфана, Елерса - Данлоса, ахондроплазія, недосконалий остеогенез, міотонічна дистрофія, хорея Гентінгтона та інші. Ознаками автосомно-рецесивного типу успадкування є: -передача хвороби „по горизонталі”, навіть при достатній кількості нащадків ознаки може не бути у дітей, але вона з’являється в онуків, -батьки зазвичай клінічно здорові, -чим більше дітей у родині, тим частіше в ній понад одну хвору дитину, -чим частіше трапляється мутагенний ген у популяції, тим частіше батьки хворих дітей є кровними родичами, -якщо хворі і чоловік, і дружина, то всі діти будуть хворими, -у шлюбі хворого зі здоровим народжуються нормальні діти (якщо здоровий не гетерозиготний носій патологічного гена), -у шлюбі хворого з носієм мутантного алеля народжується 50% хворих дітей, що імітує домінантний тип успадкування, -обидві статі уражуються однаково.
До хвороб, які успадковуються автосомно-рецесивно, належать: муковісцидоз, фенілкетонурія, галактоземія, гепатолентикулярна дегенерація (хвороба Вільсона-Коновалова), синдром Барде-Бідля (ожиріння, гіпогеніталізм, розумова відсталість, пігментна дегенерація сітківки, полідактилія), адрено-генітальний синдром, мукополісахаридози. Ознаки, гени яких розташовані не в автосомах, а в статевих хромосомах (Х і Y), називають зчепленими зі статтю. Ознаками Х-зчепленого домінантного типу успадкування є: -хворих жінок удвічі більше, ніж чоловіків; -хвора жінка передає патологічну ознаку 50% синів і 50% дочок; -хворий чоловік передає патологічну ознаку всім дочкам і не передає синам; -жінки (вони гетерозиготні) хворіють частіше менш тяжко, ніж чоловіки (вони гомозиготні). До найбільш частих хвороб, які успадковуються Х-зчеплено домінантно відносять: вітамін Д-резистентний рахіт (спадкова гіпофосфатемія), нетримання пігменту, рото-лице-пальцевий синдром, фокальна шкірна гіпоплазія. Характерні риси Х-зчепленого рецесивного типу успадкування: -хворіють тільки хлопчики; -близько 2/3 випадків „завдячують” матерям-носіям, 1/3 – новим мутаціям в Х-хромосомі матері; -у разі успадкування у хворих хлопчиків можуть бути хворі брати і дядьки по матері; -сестри хворих братів у разі успадкування мають 50% вірогідності також бути носіями патологічного алеля; -здорові чоловіки не передають хвороби, -частка випадків успадкування становить понад 2/3; -хворі чоловіки передають патологічний алель усім своїм дочкам і нікому із синів; -усі фенотипічно нормальні дочки хворих чоловіків є носіями; -у шлюбі жінки-носія з хворим чоловіком 50% дочок хворі, а 50% носії, 50% синів хворі і 50% здорові. Хвороби, які успадковуються Х-зчеплено-рецесивно: розумова відсталість із ламкою Х-хромосомою, гемофілія, м’язова дистрофія Дюшена, синдром Хантера (мукополісахаридоз ІІ типу), синдром Леша-Ніхана. Ознаками Y-зчепленого типу успадкування є: -хворіють лише хлопчики, -хворий чоловік передає патологічну ознаку (якщо не порушена фертильність) усім синам і не передає дочкам. Ознаками мітохондріальної спадковості є: -хвороба передається тільки від матері, -хворіють і хлопчики і дівчатка, -хворі чоловіки не передають хворобу. У практиці лікаря-генетика нерідко трапляються випадки, коли виявлений характер передачі захворювання не підпорядковується класичним методам успадкування. Встановлено, що деякі гени, що передаються нащадкам, несуть специфічний відбиток статі батька. Це свідчить, що деякі батьківські і материнські гени мають різні ефекти, тобто проявляються у нащадків по-різному. Це явища одержало назву геномного імпринтингу. Таким чином, деякі гени передаються дітям від одного з батьків у неактивному стані. Неактивна копія гена називається імпринтованою. Встановлено близько 30 генів, які по-різному проявляються на батьківських та материнських хромосомах. Класичним прикладом хвороб імпринтингу є спадкові синдроми Прадера-Віллі та Ангельмана, основними клінічними ознаками яких є розумова відсталість різного ступеня разом із тяжкими неврологічними порушеннями. Найбільш частою причиною синдромів є внутрішньохромосомна делеція критичного регіону (q11-q13) хромосоми 15. Ця делеція характерна для 2/3 всіх хворих. Синдром Прадера-Віллі розвивається, коли дитина успадковує делецію, яка виникла на батьківській хромосомі 15, а причиною синрому Ангельмана є делеція тієї самої ділянки на материнській хромосомі 15. Тобто виникнення цих клінічно різних синдромів залежить від хромосомної перебудови у батьків (батька та матері). Хвороби експансії (збільшення числа копій ділянок ДНК, які повторюються (повтори) в індивідумів у наступних поколіннях. Феномен експансії числа тринуклеотидних повторів (ЦГГ) був уперше виявлений при молекулярно-генетичному дослідженні синдрому Мартин-Белл (синдром ламкої Х-хромосоми). Передача відбувається від матері. Крім названого синдрому, відомі інші захворювання експансії тринуклеотидних повторів (міотонічна дистрофія (19q13.3), хорея Гентингтона (4р16.3) та деякі інші. Особливостями клінічних проявів спадкової патології є: -сімейних характер захворювання; -хронічний, рецидивний перебіг; -наявність специфічних симптомів; -численні патологічні зміни органів і систем; -природжений характер хвороби; -резистентність до найпоширеніших методів терапії. У спадкових хвороб не існує патогномонічних ознак. Частіше за все одні й ті самі симптоми спостерігаються при кількох або навіть багатьох хворобах. Наприклад, деформації хребта зустрічаються більше ніж при 50 спадкових захворювань. Семіотика спадкових хвороб вивчає ознаки (симптоми) спадкових хвороб і патологічних станів, викликаних впливом спадкових факторів і факторів середовища. У клінічній генетиці широко використовується поняття «синдром», яке вживається вже не стільки для позначення сукупності симптомів, об'єднаних одним патогенезом, скільки для позначення самостійних нозологічних одиниць. Багато нозологічно ідентифікованих спадкових хвороб називають синдромами (наприклад, хвороба чи синдром Дауна). Критерієм виділення синдромів є стійке сполучення симптомів, включаючи МАР. У генетиці використовується синдромологічний аналіз, під яким потрібно розуміти узагальнений аналіз усіх клінічних проявів з метою виявлення сталого поєднання ознак для встановлення діагнозу. При спадкових хворобах характерний розвиток хронічного процесу внаслідок постійного впливу мутантного гена. Ступінь хронізації і прогредієнтності однієї і тієї самої хвороби у різних хворих різний, що пояснюється взаємодією генів (генотип кожної людини індивідуальний). Характерним є рецидивний перебіг спадкової патології, зумовлений як генетичними факторами, так і факторами середовища. До генетичних факторів відносять особливості функціонування генів хворого, тобто регуляцію їх активності в межах, установлених генотипом. Фактори середовища ускладнюють основний патологічний процес (активізація мікробного фактора, порушення харчування) та спричиняють додатковий пошкоджувальний вплив. Необхідно хворого оглядати повністю. Наприклад, у хворого з вродженою вадою серця потрібно уважно оглянути руки: вкорочення 1 пальця кисті чи наявність трьох фаланг замість 2 наводить на думку про домінантно успакований синдром Холт-Орама (синдром “рука-серце”). Розумова відсталість - результат патології більше ніж 100 спадкових синдромів.
Дата добавления: 2015-12-16 | Просмотры: 845 | Нарушение авторских прав |