|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
КОРОТКИЙ ВИКЛАД НАВЧАЛЬНОГО МАТЕРІАЛУ. Патогенез метаболічних хвороб можна уявити як появу мутантної алелі, що призводить до недостатності
Патогенез метаболічних хвороб можна уявити як появу мутантної алелі, що призводить до недостатності, відсутності, аномального чи надмірного продукту (фермента). Це порушує ланцюг біохімічних процесів у клітинах організму, в органах і з часом призводить до патологічних проявів в організмі. Організм людини при накопиченні продукту в результаті змін біохімічних реакцій намагається “відшукати” альтернативні шляхи переробки цього продукту. При цьому часто створюються продукти, які є токсичними для організму. Значна кількість метаболічних хвороб (МХ) проявляється в неональному періоді. Акушерський анамнез допомагає запідозрити МХ (спонтанні аборти чи мертвонародження як елімінація нежиттєздатної дитини, чоловіча стать може свідчити про Х-зчеплену форму). Патологічний перебіг вагітності (тривале блювання, жирова дистрофія печінки) можуть бути наслідком порушення у плода окислення жирних кислот. Гіперактивність плода може бути як наслідок порушення метаболізму та судом, які виникають внутрішньоутробно. Основними проявами МХ у період новонародженості є відмова від їжі, втрата маси тіла, гіпотонія, гепатомегалія, летаргія, порушення дихання. Потім з’являються судоми, кома, поліорганні зміни. Частіше ці симптоми поєднуються з інфекціями і захворюванням, що має прогресуючий перебіг. Як результат несприятливого впливу метаболічних хвороб у період внутрішньоутробного розвитку в новонародженого можуть бути дисморфічні риси обличчя, формуються фенотипи синдромів (Целльвегера, Рефсума, Сміта-Лемлі-Опіца). Накопичення продукту в результаті патологічного обміну спричиняє формування специфічного зовнішнього вигляду (синдром Гурлера внаслідок порушення розпаду мукополісахаридів, Тея-Сакса внаслідок порушення розпаду гангліозидів). Через порушення обміну у новонароджених уже з перших годин життя можуть розвинутись інтоксикація, енцефалопатія (при порушенні циклу сечовини), при органічних ацидуріях (блок обміну амінокислот) чи в результаті блоку в обміні галактози (галактоземія). Така інтоксикація має затяжний характер і призводить до декомпенсації. Сприяють виникненню декомпенсації метаболічних хвороб голодування, інфекції, операції та травми. Ці фактори впливають на хворих як з порушеннями білкового, так і вуглеводного та енергетичного обміну. При надлишку споживання білків у дітей з МХ можуть ініціюватися прояви аміноацидопатій, органічних ацидурій, дефекту обміну сечовини. Вуглеводне навантаження у дітей з МХ сприяє проявам мітохондропатій. Велика кількість спожитих фруктів сприяє проявам непереносимості фруктози, а молочних продуктів – галактоземії. Значне жирове навантаження викликає маніфестацію порушення жирних кислот. Зловживання деякими ліками призводить до маніфестації порфірії, а фізичні навантаження провокують гемолітичний криз у разі мітохондропатій. Метаболічні хвороби у різних вікових групах можна запідозрити у таких випадках (Гречаніна О.Я. та співавт., 2007): - невстановлена причина раптової смерті; - незвичайний запах видихуваного повітря, тіла, сечі, незвичайний колір сечі; - порушення психо-фізичного розвитку; - наявність сибсів з невстановленим діагнозом (сепсисом, енцефалопатією); - поява ознак хвороби при зміні дієти; - пристрасть чи відраза до окремих продуктів; - судоми; - порушення м’язового тонусу; - органомегалія; - ураження шкіри (попрілості, пігментації, товста шкіра); - обмеження рухомості суглобів; - часті блювання та гикавка; - гірсутизм; - кровноспоріднений шлюб. Стійкі судоми в період новонародженості за відсутності в анамнезі асфіксії під час пологів є однією з перших ознак МХ. Причинами судом при цьому можуть бути також: недостатність піридоксину, некетотична гіпергліцинемія, дефіцит 3-фосфат-гліцератдегідрогенази, дефіцит біотинідази, дефіцит сульфітооксидази, порушення пуринового обміну, пероксисомні хвороби. При діагностиці метаболічних хвороб виникають труднощі, які пов’язані з особливостями перебігу, маскуванням проявів під інші захворювання (перинатальні енцефалопатії, пологові травми, сепсис, імунодефіцит, інфекції).
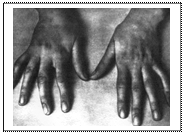
Згідно із сучасною класифікацією метаболічних хвороб (Zschoke J., Hoffman G. (1999) виділяють: 1. Порушення проміжного метаболізму 1.1. Порушення метаболізму амінокислот (аміноацидопатії): - органічні ацидурії; - порушення детоксикації аміаку. 1.2. Порушення окислення жирних кислот та ектогенезу. 1.3. Порушення вуглеводів і їх транспортування: - порушення метаболізму галактози і фруктози; - порушення метаболізму гліцерину; - порушення глікогенезу і накопичення глікогену; - порушення транспортування вуглеводів. 1.4. Мітохондріальні порушення. 1.5. Порушення, пов’язані з недостатністю вітамінів. 1.6. Порушення транспортування амінокислот. 1.7 Порушення метаболізму пептидів. 1.8. Порушення метаболізму мінералів (міді, заліза, цинку). 2. Порушення біосинтезу і розщеплення складних молекул 2.1. Порушення метаболізму пуринів і піримідинів. 2.2. Лізосомні хвороби накопичення. 2.3. Порушення метаболізму ізопреноїдів і стеринів. 2.4. Порушення метаболізму жовчних кислот і гемів. 2.5.Уроджені порушення глікозилювання. 2.6. Порушення метаболізму ліпопротеїнів. 3. Дефекти медіаторів і пов’язані з ними порушення 3.1. Порушення метаболізму гліцину і серину. 3.2. Порушення обміну стеринів і біогенних амінів. 3.3. Порушення метаболізму епсилон-амінобутиратів. Більшість метаболічних хвороб успадковується автосомно-рецесивно, що важливо, якщо родина має обмежений етнічний чи географічний фенотип. Частіше підозра на МХ з’являється при народженні другої дитини і появою симптомів хвороби, від якої померла перша дитина (сепсис, енцефалопатія). Відомо, що клінічні прояви МХ навіть у членів однієї сім’ї можуть значно варіювати. Варто значну увагу приділити анамнезу, адже жінки можуть не пам’ятати, від якої хвороби лікувалися, а тільки, що була особлива дієта (наприклад, при фенілкетонурії). Вік маніфестації МХ може бути різним, хоча частіше прояви патології проявляються в перші дні життя (наприклад, хвороба “кленового сиропу” маніфестує на 5-7-й день життя). Період ризику – це друге півріччя життя, коли дитина починає вживати різноманітну їжу. Хвороби, високодозована стероїдна терапія, травми, голодування можуть провокувати порушення у будь-якому віці. Для встановлення діагнозу під час обстеження пробанда з метаболічними порушеннями необхідно дотримуватися стандартизованої класифікації основних характеристик. Синдром Марфана –це спадкова патологія сполучної тканини. Описаний Марфаном В. у 1886 році. Причина – мутація в гені фібриліну (15-а хромосома). Автосомно-домінантний тип успадкування. В 75% хворі успадковують патологію від батька. У хворих з цим синдромом порушується утворення поперечних зв’язків колагену та утворення колагенових пучків, збільшена розчинність колагену. Основними проявами синдрому Марфана є: - після народження дитини помітне переважання довжини тіла над масою. У період росту дитини спостерігається видовження обличчя, кінцівок (арахнодактилія), відбувається деформація скелета у вигляді вираженого сколіозу, лійкоподібної грудної клітки; - зміни в серцево-судинній системі спочатку розцінюються як пролапс мітрального клапана, але до 40 років часто діагностується аневризма аорти, яка розшаровується і часто закінчується летально; - у периферійних судинах утворюються аневризми, часто наявні аортальна регургітація, застійні серцеві порушення та порушення ритму; - часто у цих хворих зменшення розміру кришталика (мікрофакія), кришталик круглої форми (сферофакія), що призводить до підвивиху кришталика. Крім міопії та підвивиху кришталика, спостерігаються відшарування сітківки, сплощення рогівки; -у дітей утворюються грижі та атрофічні стриї; - може спостерігатися спонтанний пневмоторакс;
Арахнодактилія При лабораторному обстеженні відмічається збільшення в 2 рази екскреції глікозаміногліканів. Значно зростають показники (удвічі й більше) оксипроліну (величини залежать від тяжкості захворювання). У хворих також в 5-10 разів підвищений рівень соматотропного гормону. З інструментальних методів обстеження використовують рентгенографію хребта (виявлення та оцінка ступеня сколіозу), обстеження з допомогою щілинної лампи для встановлення підвивиху кришталика, УЗД серця допомагає виявити дилятацію аорти та дегенеративні зміни клапанів серця. У лікуванні синдрому використовується обмеження фізичної активності. Призначення пропранолону (анапреліну) може сповільнити розшарування аорти. Диспансеризація хворих проводиться дільничним лікарем, кардіологом, офтальмологом, хірургом-ортопедом, генетиком, психологом. При показаннях – хірургічна корекція серцево-судинної системи.
Причинами виникнення кровотеч при синдромі Елерса-Данлоса є аномальність колагену, що порушує еластичність судин, порушення агрегації тромбоцитів та зниження їх адгезивності. Клінічними критеріями синдрому є: - пасивне згинання надп’ятково-гомілкових суглобів на 90о; - торкання великим пальцем кисті передпліччя; - перерозгинання у ліктьовому суглобі понад 10о;
- гіперрозтяжність шкіри, відшарування сітківки, множинний карієс, грижі, спонтанні перфорації кишківника, пролапс мітрального клапана, птоз внутрішніх органів, внутрішньочерепні крово-виливи. Лікування – симптоматичне. Гіперрозтяжність шкіри при синдромі Елерса-Данлоса
Гіпертрофія клітора та зростання губно-мошонкової складки Це найбільш часта форма гіперплазії наднирників. Дефіцит 21-гідроксилази призводить до порушення утворення дезоксикортикостерону та 11-дезоксикортизолу. Біосинтез андрогенів не порушений, що призводить до надлишкової їх продукції ще в період внутрішньоутробного розвитку. У новонароджених дівчаток відмічається різний ступінь маскулінізації від помірної гіпертрофії клітора до повного зростання губно-мошонкової складки з формуванням передміхурової залози, мошонки, статевого члена з отвором сечовидільного каналу. Внутрішні статеві органи сформовані правильно (каріотип 46.ХХ). Відмічається гіперпігментація біля соскової та генітальноїділянок. У хлопчиків основними клінічними симптомами є ранній статевий розвиток та низький зріст, пов’язаний з передчасним закриттям зон росту. При неповному дефіциті 21–гідроксилази електролітний баланс, рівень кортизолу та альдостерону в нормі. При повному дефіциті фермента (сільвтрачальна форма) – на перший план виступають симптоми блювання, тахікардії, сонливості, ознак дегідратації. Необхідно диференціювати з уродженим пілоростенозом; - адреногенітальний синдром з артеріальною гіпертензією (дефіцит 11в-гідроксилази). При цьомупорушується перетворення 1-дезоксикортикостролу в кортизол і дезоксикортикостерону в кортикостерон. Клінічно це проявляється верилізацією і в деяких випадках гіпертонією. У дівчаток відмічається гіпертрофія клітора, зростання губно-мошонкової складки з утворенням мошонки. Формування внутрішніх статевих органів не порушене. У хлопчиків відмічена пігментація мошонки. Лабораторно виявляється підвищена екскреція з сечею 17-оксикортикостероїдів та 17-гідроксикортикостероїдів, зниження рівня кортикостеролу й альдостеролy в крові. Лікування – замісна терапія гормонами. Факоматози – спадкові захворювання, які характеризуються одночасним ураженням нервової системи та шкіри. У нашій державі вперше описав їх в 1932 році Давиденков С.Н. і назвав факоматозами. При цьому виділив дві групи: ангіоматозні та бластоматозні форми. Враховуючи те, що факоматози часто супроводжуються з пухлинами, деякі дослідники відносять їх до гамартозів. Нині вдалося картувати гени, які відповідають за розвиток нейрофіброматозу, туберозного склерозу, синдрому Луї-Бар, хвороби Гіппеля-Ліндау, синдрому неутримування пігменту, синдрому базально-клітинного невуса та ін.
1. Бластоматозні форми: а)нейрофіброматоз, б) туберозний склероз. 2. Ангіоматозні форми факоматозів: а) хвороба Штурне-Вебера; б) хвороба Гіппеля-Линдау; в)хвороба Рандю-Ослера; д) синдром Кліппеля-Треноне; е) синдром Луї-Бар. 3. Раритетні форми факоматозів: а) гемангіоматоз розсіяний; б) дисплазія ектодермальна гідротична; в) невус сальних залоз лінійний; г) нейрошкіряний меланоз; д) ангіо-матоз дифузний кортико-менінгеальний Діврі Богаерта; е) синдром неутримування пігменту; є) синдром Лешке; ж) синдром базально-клітинного невуса; з) гемангіоматоз печінки; і) синдром Маффучі; к) синдром множинного лентиго. 4. Некласифіковані форми.
Частота фако-матозів у різних регіонах досить
Характерною особливістю фако-матозів є значне розмаїття клінічних проявів.
Малігнізація нейрофіброми 1-го типу Так, основними клінічними ознаками нейрофіброматозу є пухлинне утворення типу нейрофібром, неврином периферійних, шкірних та черепно-мозкових нервів, а також шкірні пігментації (плями кавового кольору, голубі невуси, пігментні родимки тощо). При нейрофіброматозі часто зустрічаються доброякісні новоутворення - невриноми, фіброми, ліпоми, ангіоміоліпоми, фіброміоми. Неврологічна симптоматика при нейрофіброматозі проявляться у вигляді частих судомних пароксизмів, головних болів, лікворно-гіпертензійного синдрому, зниження слуху, зору, вогнищевої неврологічної симптоматики, зниження інтелекту різного ступеня, пухлинних уражень периферичних нервів та нервів шкіри. Спостерігаються різноманітні ураження вегетативної нервової системи. Вирізняють два типи нейрофіброматозу. Перший включає кожні прояви у вигляді плям кави з молоком, вузликів Ліша, нейрофібром, скелетних порушень, розумової відсталості, підвищення ризику малігнізації, частота якої сягає 5,2%. Діагностичними критеріями є наявність шести та більше
Другий тип супроводжується пухлинним ураженням восьмої пари черепно-мозкових нервів та іншими внутрішньочерепними та інраспінальними утвореннями. Ця форма характеризується наявністю множинних неврином черепно-мозкових нервів та корінців спинного мозку, а також менінгом та гліом церебральної та спіральної локалізації. Діагностичними критеріями є: збільшення маси 8-ї пари черепно-мозкових нервів, що визначається ядерною магнітно-резонансною томографією (ЯМРТ) та залежність між нейрофібромою та пухлиною слухового нерва (нейрофібромою, менінгіомою, гліомою, шванномою). Симптоматика при цьому залежить від локалізації пухлини: зниження слуху, зору, цефалгій, лікворно-гіпертензійні та епілептиформні синдроми. При обстеженні за допомогою ЯМРТ часто виявляються пухлини іншої локалізації. Туберозний склероз є одним із варіантів бластоматозних факоматозів. При ньому нашкірні зміни зустрічаються в 60-90% хворих у вигляді аденом сальних залоз Прігла, депігментованих плям, пухлин шкіри, підшкірних фібром, папілом в області ясен, ліпом, ангіофіброліпом, множинних пігментних плям, ангіом, ділянок “шагреневої шкіри”, підшкірних навколонігтьових фібром, ангіофібром. Аденоми сальних залоз є найбільш характерною ознакою туберозного склерозу і проявляються у вигляді жовтих чи рожевих утворень діаметром 1-3 мм, одиноких чи множинних, розташованих у вигляді метелика на щоках, підборідді, носо-губних складках, на шкірі повік, вушних роковин, появляються у віці від 3 до 13 років. Рідко спостерігаються у дітей до 2 років. Найбільш типовими ознаками ураження нервово-психічної сфери при туберозному склерозі є судоми (62-80%) та розумова відсталість (38-50%). Майже в 40% наявні офтальмологічні ураження (Гречаніна О.Я., Молодан Л.В., 2003). На диску зорового нерва наявні утворення у вигляді білуватих чи сіруватих ділянок. Можуть спостерігатися пухлини, плями жовто-червоного кольору. При цій патології часто відмічається ураження кори головного мозку, серця, спостерігаються полікістоз та ангіоміоліпоми нирок. У 98% дітей з допомогою ЯМРТ виявляться ознаки гідроцефалії. Ангіоматозні форми факоматозів (синдроми Кліппеля-Треноне, Луї-Бар, хвороби Рандю-Ослера, Гіппеля-Ліндау, Штурне-Вебера) відрізняються значним клінічним поліморфізмом. Синдром Кліппеля-Треноне маніфестує з народження у вигляді судинної родимої плями з подальшим розширенням венозних судин та гемігіпертрофією м’яких тканин і кісток. У типових випадках уражена одна із нижніх кінцівок, але можливі ураження і рук, грудей, шиї. Вроджені аномалії розвитку глибоких вен супроводжуються їх вторинним варикозом. Можливі гемігіпертрофії половини тіла, включаючи голову, шию, тулуб, кінцівки, та перехресна гіпертрофія (гіпертрофія зліва чи справа до певного рівня). Одночасно із судинними плямами у хворих можуть спостерігатися гемангіоми, плями кольору кави, невуси, ділянки депігментації шкіри, мармуровість тощо. При ЯМРТ відмічаються значне збільшення хориїдального сплетіння, тяжка церебральна атрофія, церебральні кальцифікати. Для ангіоматозу Гіппеля-Ліндау характерна класична тріада: ангіоматоз сітківки, головного мозку, кістозні утворення чи пухлини з утворенням кіст головного мозку та внутрішніх органів. Ранніми ознаками хвороби часто буває прогресуюче зниження гостроти зору, зміни на очному дні. Симптоми ураження нервової системи залежать від локалізації, і спочатку проявляються загальні симптоми (головний біль дифузного характеру, важкість у голові, відчуття тиску на очні яблука, на висоті головної болі – нудота та блювання). До ранніх ознак відносять епілептичні припадки. При втягненні в процес мозочка з’являються ознаки порушення координації, ністагм, атаксія, тремор, м’язова гіпотонія тощо. Коли наявні ураження спинного мозку, то у хворих появляються ознаки втрати чутливості, корінцеві болі. Ураження внутрішніх органів проявляться полікістозом та пухлинами (полікістоз нирок, гіпернефрома, феохромоцитома, карцинома нирок, гемангіобластоми, пухлини підшлункової залози). Для синдрому наявні 6 ознак: ангіоматоз сітківки, гемангіобластома мозочка, ангіобластома спинного мозку, гемангіобластома кісткового мозку, феохромоцитома, карцинома нирки. Наявність хоча б двох ознак дає підстави думати про це захворювання. Синдром Луї-Бар характеризується телеангіоектазіями шкіри та слизових, змінами нервово-психічної сфери (мозочковий синдром), змінами з боку внутрішніх органів у результаті вродженого дефіциту імуноглобулінів, високою частотою неоплазій. Для синдрому характерна хромосомна нестабільність (транслокації 7 та 14). Телеангіаектазії локалізуються на кон’юнктиві, шкірі щік, у міжлопатковій ділянці. Ознаки з’являються у віці 2-6 років. За рахунок гіпоміміки обличчя дітей має характерний вигляд. У дітей з’являються міоклонічні скорочення. Наявні порушення як клітинного, так і гуморального імунітету, через те у них хронічні бронхолегеневі захворювання, отити, гайморити, пієлонефрити.
3. Уроджені та спадкові захворювання нирок Обґрунтування теми. Поширеність уроджених вад розвитку сечової системи серед новонароджених досить значна і становить 18-22 на 1000 (Резник Б.Я. та співавт., 1994). Значне зростання цієї патології пояснюється широким використанням ультразвукової діагностики. Своєчасна діагностика спадкових захворювань нирок дасть змогу використовувати адекватну терапію. Дата добавления: 2015-12-16 | Просмотры: 687 | Нарушение авторских прав |
 - спостерігаються зміни ЦНС (попереково-куприкове менінгоцеле, аномалії розвитку). Кожний четвертий хворий із синдромом Марфана має розумову відсталість різного ступеня.
- спостерігаються зміни ЦНС (попереково-куприкове менінгоцеле, аномалії розвитку). Кожний четвертий хворий із синдромом Марфана має розумову відсталість різного ступеня.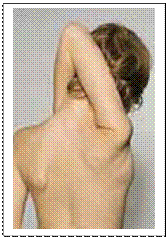 Синдром Елерса-Данлоса – це спадкова вада сполучної тканини. Характерними ознаками патології є підвищена розтяжність шкіри, ламкість судин та схильність до кровотеч. Ідентифіковано понад 10 форм цього синдрому, які з клініко-генетичного погляду можна розглядати як самостійні нозології, хоча їхні симптоми подібні.
Синдром Елерса-Данлоса – це спадкова вада сполучної тканини. Характерними ознаками патології є підвищена розтяжність шкіри, ламкість судин та схильність до кровотеч. Ідентифіковано понад 10 форм цього синдрому, які з клініко-генетичного погляду можна розглядати як самостійні нозології, хоча їхні симптоми подібні.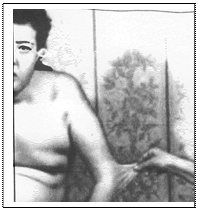 - перерозгинання у колінному суглобі понад 10о;
- перерозгинання у колінному суглобі понад 10о;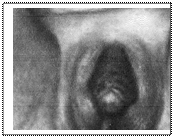 Адреногенітальний синдром – уроджена патологія, що характеризується порушенням функції наднирників. Основний тип успадкування аутосомно-рецесивний. Ген локалізації 6р21.3 (дефіцит 21-гідроксилази). Основними діагностичними ознаками захворювання є прогресуюча верилізація, пришвидшений соматичний розвиток, підвищена екскреція гормонів кори наднирників. Описано 5 типів синдрому. Найбільш часто зустрічаються: - адреногені-тальний синдром із втратою солей чи без неї (дефіцит 21-гідроксилази) або жіночий псевдогермафродитизм.
Адреногенітальний синдром – уроджена патологія, що характеризується порушенням функції наднирників. Основний тип успадкування аутосомно-рецесивний. Ген локалізації 6р21.3 (дефіцит 21-гідроксилази). Основними діагностичними ознаками захворювання є прогресуюча верилізація, пришвидшений соматичний розвиток, підвищена екскреція гормонів кори наднирників. Описано 5 типів синдрому. Найбільш часто зустрічаються: - адреногені-тальний синдром із втратою солей чи без неї (дефіцит 21-гідроксилази) або жіночий псевдогермафродитизм.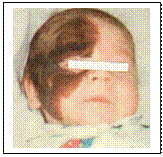 Класифікація факоматозів
Класифікація факоматозів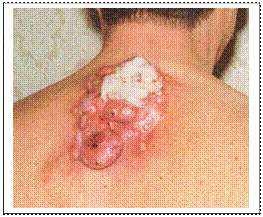 різниться і стано-вить від 1:12000 – 1:100000.
різниться і стано-вить від 1:12000 – 1:100000.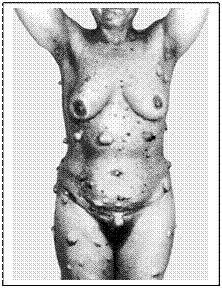 гіперпігментованих плям діаметром не менше 5 мм в осіб препубертатного віку і більше 15 мм пубертатного; дві або більше нейрофіброми одного типу або одна плексиформна нейрофіброма; наявність пігментних плям у підпахвинній чи паховій ділянках; оптична гліома; дві або більше гамартоми райдужної оболонки ока; характерні кісткові ураження типу сфероїдальної дисплазії; наявність у сім’ї такого хворого (першим типом нейрофіброматозу). Дві із перелічених ознак достатні для встановлення діагнозу. Для цієї форми характерна значна варіабельність клінічних ознак навіть серед членів однієї родини.
гіперпігментованих плям діаметром не менше 5 мм в осіб препубертатного віку і більше 15 мм пубертатного; дві або більше нейрофіброми одного типу або одна плексиформна нейрофіброма; наявність пігментних плям у підпахвинній чи паховій ділянках; оптична гліома; дві або більше гамартоми райдужної оболонки ока; характерні кісткові ураження типу сфероїдальної дисплазії; наявність у сім’ї такого хворого (першим типом нейрофіброматозу). Дві із перелічених ознак достатні для встановлення діагнозу. Для цієї форми характерна значна варіабельність клінічних ознак навіть серед членів однієї родини.