|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
КОРОТКИЙ ВИКЛАД НАВЧАЛЬНОГО МАТЕРІАЛУ
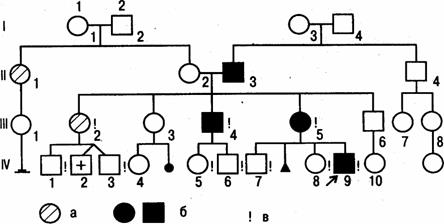
Усі спадкові хвороби поділяються на генні й хромосомні. Генні – зумовлені генними мутаціями. Хромосомні – зумовлені хромосомними та геномними мутаціями. Мультифакторіальні хвороби зумовлені комбінацією генетичних та негенетичних факторів. Крім того, виділяють генетичні хвороби соматичних клітин (пухлина Вільмса), хвороби генетичної несумісності матері й плода (гемолітична хвороба новонароджених). Хромосомні хвороби -це спадкові захворювання, які обумовлені змінами числа хромосом чи їх структури (хромосомними мутаціями). Хромосомні хвороби, як правило, не передаються нащадкам і зустрічаються як спорадичні випадки. Якщо мутація виникла в гаметах, то розвивається повна форма хвороби, а якщо на стадії поділу зиготи – мозаїчна форма хвороби. Хромосомні мутації охоплюють велику кількість генетичного матеріалу і характеризуються множинними ураженнями. Вони спричиняють близько половини випадків загибелі плода після імплантації та до 70% 2-4-х тижневих викиднів. У великих стаціонарах хворі на хромосомні захворювання займають до ¼ всього ліжкового фонду. Для цього виду патології найбільш інформативним є цитогенетичний метод дослідження. Причинами спадкових хвороб є геномні, хромосомні та генні мутації. Геномні мутації пов’язані зі зміною кількості хромосом. Хромосомні мутації пов’язані зі зміною структури хромосом, генні мутації - це молекулярні зміни на рівні ДНК. Чим більше хромосомного матеріалу залучено до мутації, тим більш неспецифічні тяжкі зміни відбуваються у фізичному і психічному розвитку людини. З удосконаленням цитогенетичних методів, особливо таких, як диференційне фарбування і молекулярна цитогенетика, відкрилися нові можливості для виявлення раніше не описаних хромосомних синдромів і встановлення зв’язку між каріотипом і фенотипом при невеликих змінах хромосом. Цитогенетика використовується не тільки для діагностики хромосомних захворювань, але і для діагностики патології внутрішньоутробного розвитку (спонтанні аборти, викидні), лейкозів. Число описаних хромосомних хвороб наближається до 1000, із них більше 100 має чітку клінічну картину і називається синдромами. Близько половини всіх зачать не має подальшого розвитку ще в перші дні до встановлення факту вагітності. Народжується тільки кожна десята дитина, яка мала хромосомні порушення. Процес зміни генетичного матеріалу одержав назву „мутагенез”. Вирізняють спонтанний та індукований мутагенез. Індукований – це мутагенез, що викликається несприятливим впливом навколишнього середовища (іонізувальне опромінення, фізичні, хімічні та біологічні фактори (віруси)). Діагностика хромосомних аномалій необхідна в практиці лікаря різних спеціальностей (генетика, акушера-гінеколога, педіатра, невролога, ендокринолога тощо). В розвинених країнах у багатопрофільних лікарнях (більше 1000 ліжок) наявні цитогенетичні лабораторії. Методи дослідження, які використовуються для діагностики в медичній генетиці: генеалогічний, популяційно-статистичний, близнюковий, методи дерматогліфіки і пальмоскопії, біохімічні, електрофізіологичні методи, цитогенетичний метод. Генеалогічний метод - метод родоводів, вивчає закономірності передачі спадкових ознак індивіда в ряді послідовних поколінь. Метод ґрунтується на складанні й аналізі родоводів. Людина, що обстежується і родовід якої складають, називається пробандом (пропозитом). Пробанд – хворий чи носій ознаки, що вивчається. Дітей однієї батьківської пари називають сибсами (брати – сестри). Сім’я – батьки та їх діти, а якщо включають кровних родичів – рід. Клініко-генеалогічний метод дослідження – це вивчення родоводу та поширення патологічної ознаки в родині (роді) за допомогою клінічного обстеження зі вказанням родовідних зв’язків між членами сім’ї (роду). Складовими генеалогічного аналізу є встановлення спадкового характеру ознаки та типу спадкування. Метод застосовують для: -встановлення спадкового характеру ознаки; -визначення типу успадковування, пенетрантності гена; -аналізу зчеплення генів і картування хромосом; -вивчення інтенсивності мутаційного процесу; -розшифровування механізмів взаємодії генів; -медико-генетичного консультування. Спадковий характер досліджуваної патологічної ознаки (хвороби) можна запідозрити, якщо вона кілька разів трапляється у родоводі. Однак можливі фенокопії - вплив того самого патогенного фактора на кількох членів родини. При складанні родоводу керуються правилами: -пробанда на схемі родоводу позначають стрілкою; -особи одного покоління займають окремий рядок або коло; -покоління позначають зліва римською цифрою (найстарше цифрою 1, а наймолодше – внизу родоводу); -усіх членів одного покоління розміщують у порядку народження (зліва направо) по горизонталі й позначають арабськими цифрами; -до родоводу включають усіх членів сім’ї. При посиланні на будь-якого члена сім’ї спочатку вказують номер покоління, а потім номер члена (наприклад ІІ-3). До схеми родоводу додається легенда (систематизовані дані про пробанда та його родичів). У легенді відмічаються дані обстеження пробанда, відомості про огляд родичів, зіставлення результатів огляду пробанда з даними опитування його родичів, з письмовими відомостями про родичів, що мешкають в іншій місцевості. Проведений аналіз родоводу дозволяє встановити, чи є дана ознака (хвороба) спадковою (чи досліджувана ознака зустрічається в родоводі кілька разів), тип успадкування (домінантний чи рецесивний, автономний чи зчеплений зі статтю, зиготність пробанда (гомозигота чи гетерозигота) за досліджуваною ознакою, встановлюється ймовірність ризику прояву спадкової ознаки в нащадків.  - особа чоловічої статі -стать невідома - особа чоловічої статі -стать невідома
 
  - монозиготні близнюки - монозиготні близнюки
 
-викидень -аборт -мертвонароджений

домінантного гена
  -померлі - пробанд
Рисунок 1.6 – Символи, які використовують при складанні родоводу (Г.Юст, 1931 р.)
Рисунок 1.7 - Приклад родоводу. Позначення стандартні: а - хворі на діабет; б - хворі на нейрофіброматоз; в - особисто обстежені
Метод б лизнюків ґрунтується на схожості й різниці монозиготних і дизиготних близнюків. Близнюків на планеті 1,5-2%. Монозиготні близнюки виникають із однієї яйцеклітини, а дизиготні – із різних. Помічена спадкова схильність до народження близнюків (частіше передається по материнській лінії). Встановлено, що близнюки частіше народжуються у жінок старшого віку (частота зростає до 37 років, а далі – знижується). Частота народження близнюків 1:86-88, серед яких ¼ монозиготні. У монозиготних близнюків відбитки пальців подібні, близнюки однієї статі, дуже подібні, мають однакові групи крові за різними системами (АВ0, MN, S, резус-фактора, в тому числі групи еритроцитарної системи: Келл, Льюїс, Лютеран, Дафі, Кід, Дієго та ін.). Між монозиготними близнюками можлива трансплантація тканин. За допомогою близнюкового методу можна вивчити роль спадковості та вплив зовнішнього середовища на формування фізіологічних та патологічних особливостей організму, конкретні фактори, які посилюють чи послабляють вплив зовнішнього середовища, кореляцію ознак та функцій. На основі близнюкового методу встановлена генетична схильність до різних захворювань (включаючи інфекційні), тривалість життя теж визначається спадковістю (на 30-40%). Встановлено збіг (конкордантність) у монозиготних близнюків за коефіцієнтом інтелектуальності (індекс ІQ), ряду особливостей психіки, схильності до асоціальних вчинків (70%). Для правильних висновків одночасно потрібно вивчати як монозиготних, так і дизиготних близнюків. Популяційно-статистичний метод широко використовується в клінічній генетиці. При цьому встановлюється частота тих чи інших вад розвитку, проводиться розрахунок ризику при деяких мультифакторіальних захворюваннях. Статистичні розрахунки можуть торкатися різних факторів ризику. Наприклад, зіставляючи дату народження дитини з вадою розвитку, датою зачаття, одержуємо можливість у ряді випадків встановити етіологічний зв’язок патології з впливом зовнішнього середовища (наприклад, підвищеною онсоляцією, захворюванням ГРВІ). Метод використовується різними розділами генетики, в тому числі для вирішення питань соціальної гігієни, для розроблення рекомедацій з профілактики погіршення генофонду попляцій. Метод дерматогліфіки (вивчення гребеневих візерунків пальців рук) використовується як допоміжний метод при діагностиці деяких хромосомних синдромів (трисомій 13, 18, 21, частково трисомій 9). Це специфічний метод клінічного обстеження, при якому вивчаються візерунки відбитків долонь та стоп. У кожного народу, у кожної раси малюнки на кінчиках пальців мають свої особливості. Узори на пальцях, на долонях індивідуальні. Метод доцільно використовувати для ідентифікації монозиготних близнюків. Цитогенетичний метод дозволяє вивчити хромосоми клітин людини, їх числові і структурні порушення. Найбільш доступний скринінг – визначення статевого хроматину (тілець Барра). У жінок з нормальним каріотипом наявне тільце Барра, а при ХХХ збільшується кількість тілець. Зазвичай статевий хроматин визначається в клітинах зіскобу слизової оболонки порожнини рота. Метод простий у виконанні та доступний. Дослідження хромосомного набору (каріологічний аналіз) проводиться в метафазних пластинках лімфоцитів (або фібробластів), які культивуються на штучних середовищах. Матеріалом для культивування є кров (лімфоцити), шматочки шкіри для культивування фібробластів. При цьому використовується як звичайне фарбування, так і попередня трипсонізація за методом М.Seabright (1972) для одержання диференційованої окресленості хромосом. За допомогою диференційного пофарбування хромосом ідентифікують структурні хромосомні аномалії (делеції, транс-локації, інверсії), виявлені рутинним методом пофарбування. Існують чотири основних методи диференційного пофарбування хромосом: Q-, G-, R- та С- методи. У кожного з них є кілька модифікацій з огляду на техніку виконання. Структури, які виявляють по довжині хромосом, рекомендовано відповідно до типу пофарбування називати Q-, G-, R- та С-сегментами. Q-сегменти - ділянки, які флуоресціюють після забарвлення акрихіном оренжевим. G-сегменти забарвлені методом Гімзи. Q- та G-сегменти ідентичні, проте Q-сегменти дозволяють диференціювати Y-хромосому. R-сегменти –забарвлюються методом Гімзи після теплової денатурації. С-сегменти обмежують прицентромерні ділянки в обох хромосомах. Під час диференційного пофарбування визначають структурне диференціювання хромосом за довжиною, що виявляється чергуванням еу- та гетерохроматинових ділянок (темні та світлі смуги). Величина цих ділянок специфічна для кожної хромосоми відповідного плеча. Кожна хромосома має свій малюнок посмугованості. За допомогою С-методу пофарбування виявляють лише структурний гетерохроматин. Метод дослідження прометафазних хромосом дозволяє діагностувати мікроделеції хромосом (при синдромах Прадера-Віллі, Лангера-Гідеона). Можуть використовуватися і спеціальні цитогенетичні методи (при синдромі Мартіна-Белла) - аналіз поліморфізму довжини рестрикційних фрагментів. Використання методів молекулярної діагностики стало можливим з початку 70-х років минулого століття з відкриттям моноклональних антитіл та завдяки створенню методу гібридизації на фільтрах (1975). Були розроблені методи Нозерн- та Вестерн-блотингу, що дозволило виявляти РНК і білки. Важливим етапом діагностики стало відкриття 1983 року полімеразної ланцюгової реакції (ПЛР) та використання її у молекулярній діагностиці. Метод запропонований американцем Кері Мюлісом у 1983 р. За це відкриття вченому присуджена Нобелівська премія. Цей метод імітує процес природної реплікації ДНК. Метод складається із стадії підготовки матеріалу (виділення ДНК), ампліфікації ДНК (реплікація ДНК in vitro) та детекції результатів ампліфікації (електрофорез). Метод дозволяє протягом кількох годин виділити та розмножити певний фрагмент ДНК у кількості, що перевищує початкову в 109 разів. Використовується, наприклад, при діагностиці міодистрофії Дюшена. Виділення ДНК полягає в екстракції із матеріалу та виділенні чи нейтралізації домішок, які можуть зупиняти реакцію. Ампліфікація (природна реплікація ДНК in vitro) складаться із трьох етапів, що проходять при різних температурних режимах: -денатурація (розплітання спіралі ДНК і розходження ниток, здійснюється при температурі 93-95 градусів); -відпал (приєднання праймерів, при температурі 50-65 градусів); -синтез ДНК (комплементарне добудування обох ниток ДНК, при температурі 70-72 градуси). Матеріалом для синтезу нових ланцюгів служать дезоксирибонуклеозидтрифосфати, що додаються в розчин. Процес синтезу каталізується ферментом taq-ДНК-полімеразою. Далі цикли повторюються знову – ланцюги, що утворилися в першому циклі ампліфікації ДНК, служать матрицями для другого циклу ампліфікації. За 30-40 циклів у розчині накопичується близько 108 молекул, що достатньо для візуальної детекції цього фрагмента за допомогою методу електрофорезу в агарозному гелі. При електрофорезі в гель добавляють барвник – бромистий етидій, що дає змогу фіксувати розігнані ДНК в ультрафіолетовому діапазоні світла за допомогою фотокамери. За допомогою ДНК-діагностики є можливість вирішити такі завдання: -підтвердження клінічного діагнозу чи диференційна діагностика захворювання; -пресимптоматична діагностика, коли клінічні прояви захворювання ще на настали (пізній дебют хвороби); -діагностика носійства у фенотипово здорових гетерозиготних носіїв мутації в гені, -пренатальна діагностика по ДНК плода (амніотична рідина, хоріон, кров плода), -преімплатаційна діагностика по ДНК яйцеклітини, що дробиться (запліднена in vitro). Пресимптоматична діагностика відкриває нові можливості превентивного (попереджувального) лікування (наприклад, порушення міді при хворобі Вільсона-Коновалова). Пренатальна діагностика дітей із важкими спадковими захворюваннями (міодистофія Дюшена, муковісцидоз, міотонічна дистрофія) дозволяє сім’ям, які мають уже хвору дитину, надіятися народити здорову дитину. Жінки, носії патогенного гена (підтвердженого ДНК-діагностикою), коли вірогідність хворої дитини (наприклад, гемофілія А та В) становить 50% при проведенні пренатальної діагностики (амніоцентез, кордоцентез із подільшою ДНК-діагностикою) можуть мати можливість народжувати здорову дитину. Для масового сканування поширених мутацій використовується метод ДНК-чипів. В основі методу лежить принцип комплементарної гібридизації. Комплементарна гібридизація умовно складається з чотирьох кроків.
ДНК-чип являє собою пластинку площею близько 1 см2, на якій в чітко визначеному порядку розміщені комірчини, кожна з яких містить одноланцюгові полінуклеотиди однієї визначеної послідовності основ. При цьому розроблені методики, коли олігонуклеотиди синтезуються безпосередньо на поверхні чипа. З’явилася можливість розмістити на 1 см2 їх кілька мільйонів. Постійно здійснюється удосконалення ДНК-чипів. Розроблені та застосовуються ДНК-чипи для діагностики таласемій, муковісцидозу, раку молочної (грудної) залози, спадкової схильності до наркоманії, менінгітів та різноманітних інфекційних хвороб. Розроблені методики комбінації ДНК-чипів та полімеразних ланцюгових реакцій. Це дало можливість розширити діагностичні можливості методу та прискорити процес ідентифікації. У ДНК-діагностиці спадкових хвороб виділяють прямі та непрямі методи. При прямій ДНК-діагностиці об’єктом дослідження є мутації певного гена. Метод використовується при діагностиці муковісцидозу, фенілкетонурії, гемофілії А, міодистрофії Дюшенна, адреногенітального синдрому, таласемії, нейрофіброматозу, синдрому FRA X, недостатності альфа1-антитрипсину, недостатності 21-гідроксилази, хореї Гентингтона. Точність методу сягає 100%. Але до цього часу гени багатьох захворювань ще не повністю картовані, що знижує можливість використання методу (при адрено-генітальному синдромі, муковісцидозі, фенілкетонурії інформативність прямого методу ДНК–діагностики становить 70-80%). Непрямі методи ДНК-діагностики базуються на аналізі зчеплення з досліджуваним геном певного поліморфного локуса (маркера), з допомогою якого можна проводити маркування як мутантного, так і нормальних алелей та проаналізувати їх передачу в поколіннях (серед родичів особи, яка обстежується). Розроблені варіанти пошуку та ідентифікації точкових мутацій ДНК: метод sscр – метод аналізу конформаційного поліморфізму одноланцюгових фрагментів (реєстрація розбіжностей в електрофоретичній рухливості однониткових ДНК), метод градієнтного гель-електрофорезу (DGGE), метод хімічного розщеплення некомплементарних сайтів (СМС), метод гетеродуплексного аналізу (НА), що дає змогу ідентифікувати мутації, які перебувають у гетерозиготному стані. Використовуються також методи гібридизації нуклеїнових кислот, секвестрування ДНК (виявлення первинної нуклеотидної послідовності ДНК), метод блот-гібридизації (Едвард Саузерн, 1975), сортування хромосом за методом цитофлуорометрії. Непрямі методи аналізу ДНК більш універсальні, оскільки їх можна застосовувати і тоді, коли походження мутацій невідоме, коли ген хвороби точно не ідентифікований. Найчастіше використовується метод аналізу поліморфізму довжини ристрикційних фрагментів (ПДРФ-аналіз). Цей метод складається із етапів: -виділення геномної ДНК; -рестрикція за допомогою специфічних ендонуклеаз; -розділення фрагментів електрофорезом; -ідентифікація фрагментів за допомогою блот-гібридизації за Саузерном. Ураховуючи велику трудозатратність цитогенетичного аналізу, показаннями для таких досліджень є: -у хворих у перші місяці життя: пренатальна гіпоплазія (гіпотрофія), у поєднанні з високим рівнем стигматизації та грубими вадами розвитку чи без них; -у більш старшому віці: виражені порушення дерматогліфічної гістограми у хворих з грубою затримкою психомоторного та фізичного розвитку; -обстеження батьків, які мають дітей із хромосомною патологією; -підтвердження хромосомної хвороби, наявність якої підозрюється за клінічними ознаками; -викидні, мертвонародження, народження дітей з природженими вадами розвитку; -порушення репродуктивної функції у жінок та чоловіків; -гіпогонадизм, порушення статевого диференціювання; -синдроми, які характеризуються хромосомною нестабільністю; -лейкози, пухлинні ураження тощо. Таким чином, непряма ДНК-діагностика проводиться у випадках: 1) коли ген не ідентифікований, а лише картований на певній хромосомі, 2) коли методи прямої діагностики не дають результату (внаслідок великої протяжності гена чи великого спектра мутаційних змін), при складній екзонно-інтронній організації гена. Для непрямої діагностики можуть використовуватися гіперваріабельні сателітні повтори (метод більш інформативний, ніж ПДРФ). Використання цих повторів може використовуватися для діагностики більшості моногенних захворювань. Молекулярно-генетична діагностика спадкових хвороб використовується і для вивчення геному людини. Щоб виявити для цього необхідні специфічні фрагменти ДНК використовують блот-гібридизацію за Саузерном. Щоб виявити потрібні фрагменти, здійснюють гібридизацію ДНК-зондом або клонування фрагментом ДНК. Результат гібридизації комплементарних ланцюгів радіоактивного фрагмента ДНК знаходять за допомогою радіоавтографії (ДНК проявляється у вигляді радіоактивної смуги). За допомогою методу Саузерна можна скласти карту геному на ділянці досліджуваного гена та встановити, наявний якийсь дефект чи ні. На сьогодні є різні методи встановлення мутацій. Прямий спосіб передбачає ряд методів: -визначення нуклеїдної послідовності (секвестрування), що дає можливість виявити заміни основ, делеції, вставки у фрагменті, який вивчається; -визначення порушень місця рестрикції за допомогою блот-гібридизації за Саузерном; -проведення алелоспецифічної гібридизації із синтетичними зондами (виявлення мутацій у геном ній ДНК); -хімічне та ферментативне розщеплення ДНК у місцях неправильної зшивки основ дозволяє виявити велику групу мутацій, які призводять до нестабільності ДНК; -рестрикція змін електрофоретичної рухливості мутантних молекул ДНК; -трансляція білкового продукту здійснюється in vitro на основі одержання специфічної матричної РНК з добавлянням лузату ретикулоцитів. Синтезований білок аналізується з допомогою електрофорезу. Одним із типів поліморфізму ДНК є мікросателіти (1-, 2-, 3-, тетрануклеарні тандемні послідовності ДНК, які повторюються. Наприклад, хорея Гентигтона, багаторазово повторюється ЦАГ (цитозин-аленин-гуанін) геномної ДНК, тобно мутація гена складається з експансії (багаторазове збільшення числа копій). Подібне явище характерне і для міотонічної дистрофії, синдрому ламкої Х-хромосоми. Для вирішення питання ДНК-діагностики пацієнти направляються в Інститут медичної генетики (Харків). Показаннями для проведення пренатальних цитогенетичних досліджень є: - вік жінки до 17 та понад 35 років; - наявність хромосомної патології в одного з батьків; - народження попередньої дитини із хромосомною патологією; - результати УЗД, що припускають хромосомну хворобу плода; - низький рівень альфа-фетопротеїну (АФП) у сироватці крові вагітної; - тератогенний вплив на плід; - спонтанні викидні на ранніх термінах, - безпліддя в анамнезі; - захворювання, зчеплені зі статтю. Завдяки розвитку біології та медицини більш досконалим методом діагностики спадкових порушень обміну є біохімічний. Цей метод дозволяє провести діагностику більш ніж 1000 спадкових захворювань обміну речовин (мукополісахаридози, хвороба Марфана, синдроми Тея-Сакса, Німана-Піка, Лоуренса-Муна-Барде-Бідла та ін.). При використанні біохімічних методів важливе значення має якомога раніше обстеження. Дефекти обміну ферментів визначають за продуктами метаболізму цих ферментів у біологічних рідинах хворого. Матеріалом для дослідження є сеча, кров, ліквор та ін. Для діагностики спадкових хвороб обміну використовують двохетапну систему біохімічних досліджень. Перший етап – скринінг. При цьому, крім біохімічного, використовують мікробіологічні тести Гатрі, в основі яких лежить феномен вибіркового росту бактерій за наявності тієї чи іншої амінокислоти. Програма масового скринінгу повинна передбачати: 1. Інформування та навчання населення, лікарів і медичного персоналу. 2. Діяльність централізованої лабораторії з ефективною діагностикою. 3. Відповідна організація та постійне проведення пост-скринінгового етапу (повторне взяття крові для підтвердження первинного позитивного тесту. 4. Можливість адекватного лікування. Другий етап – це кількісне визначення вмісту метаболітів та активності ферментів у біологічних рідинах та тканинах. З 1981 року групою Ф.Сенґера розшифрована нуклеотидна структура ДНК мітохондрій (мтДНК). Для мтДНК характерні особливості: швидкі темпи мутування, компактність і відсутність інтронів, велика кількість копій у кожній клітині, материнський характер успадкування, відсутність рекомбінацій, в одній клітині можуть співіснувати одночасно нормальні й мутантні мтДНК (гетероплазмія). Встановлена кореляція між мітохондріальним генофондом і географічним проживанням особи. Роботою Алана Уїлсона, Ребекка Кенн і Марка Стоукінга в 1987 році на основі ДНК мітохондрій було зроблено висновок, що всі люди, які зараз живуть, генетично пов’язані з однією жінкою, яка жила близько 200 тисяч років тому в Африці. Дослідження показали, що людство походить від єдиного генетичного пращура (поширився близько 200 тисяч років тому). Ця гіпотеза отримала назву “генетичної” або “африканської Єви”. Сучасна людина вперше з’явилася в Південно-Західній Африці, в районі, де тепер проходить кордон між Намібією та Південно-Африканською Республікою. Такий висновок зроблено ученими університету Пенсільванії (США). Дослідження Дугласа Уолласа із університету Емері в Атланті дозволили зробити висновок, що в „африканської Єви” було 18 нащадків, кожен із певним мітохондріальним геномом, які розсіялися по всіх регіонах земної кулі (Е.Уїллет, 2008). Професор Сара Тішкоф (2009), керівник цих робіт, відзначила, що сучасне населення Африки має найбільш різноманітний генофонд. Вихід перших людей з Африки в районі Червоного моря відбувся близько 60 тисяч років тому. На думку Зінобіа Ждейкоба (2009) із університету Волога в Австралії, мігрувати людей примусили не кліматичні зміни, а ідеї та технології. На підставі генетичних відстаней встановлено, що генетичне виділення негроїдів відбулося близько 10 тис. років тому, поділ монголоїдів і європеоїдів – близько 50 тис. років тому. В корінного населення Америки встановлений азійський компонент, і його походження є генетично близьким народам Сибіру. В останні кілька тисяч років відмінності між расами зростають. Людські раси відділяються одна від одної, як заявив керівник досліджень Генрі Харпендінг (2009), професор антропології університету штату Юта (США). Дослідження показують, що генетичних відмінностей у людей в чотири рази менше, ніж у шимпанзе. Це свідчить про те, що в минулому людство пережило сильне, але короткочасне скорочення популяції до кількох тисяч або тривале, але біль помірне скорочення до десятків чи сотень тисяч осіб. Встановлено, що популяція українців належить до субкластеру, генетично подібного до популяцій сербів, німців, молдаван, угорців, хорватів і чехів. Даний субкластер поєднує популяції Центральної і Східної Європи, пращури яких вийшли із Азії, Причорномор’я, а раніше із регіонів глибинної Азії (пращури угорців хунну), мігрували азійськими і південноросійськими степами до Європи. Результати кластеризації мтДНК української популяції свідчать, що більшість типів характерні для європейської популяції. Однак серед них виявлений також азійський компонент. За даними результатів колективу російських дослідників медико-генетичного наукового центру РАМН, які працювали спільно із естонськими та англійськими колегами (2010) в центральних та південних районах Росії, населення генетично близьке до українців, білорусів та поляків, чого не можна сказати про північні та східні регіони (обстежено 1228 громадян із 14 районів). Вчені дійшли висновку, що через 100 тисяч років люди будуть високими, смаглявими, можуть втратити соціальні навики (любов, симпатії, довіру, повагу). Мутації генів мтДНК лежать в основі мітохондріальних міопатій і спадкової нейропатії Вебера. Для діагностики цих захворювань використовують біоптат скелетних м’язів. Пренатальна діагностика мітохондріальних захворювань поки не рекомендується через недостатню її інформативність. При обстеженні хворих, крім клінічних, широко використовуються також інструментальні методи дослідження. Серед них найбільш доступними є: електрофізіологічні (ЕКГ, ЕЕГ, ФКГ, міограма та ін.), рентгенологічні (оглядові, прицільні, з контрастуванням, комп’ютерна томографія), ультразвукові (сканування, доплерографія), ендоскопічні обстеження (бронхоскопія, фіброгастроскопія, ректоромано- та колоноскопія, цистоскопія), обстеження на основі використання ядерно-магнітного резонансу (МРТ).
Дата добавления: 2015-12-16 | Просмотры: 960 | Нарушение авторских прав |