|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
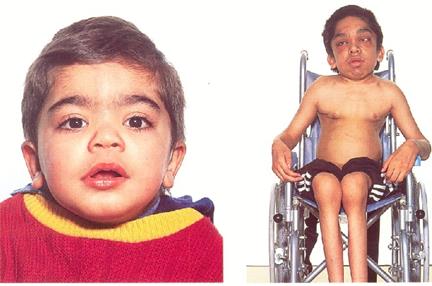
КОРОТКИЙ ВИКЛАД НАВЧАЛЬНОГО МАТЕРІАЛУ. Лізосомні хвороби - це важкі спадкові захворювання, які виникають унаслідок зниження специфічних ферментів лізосом
Лізосомні хвороби - це важкі спадкові захворювання, які виникають унаслідок зниження специфічних ферментів лізосом. Ферменти беруть участь у певному етапі обміну компонентів білків, вуглеводів та ліпідів. У результаті недостатності ферментів (активність лізосомних ферментів не перевищує 10-20% від норми) спостерігається накопичення субстрату (хвороби накопичення – тизауринози). При цьому порушується функція клітин різних тканин організму, і, як наслідок, появляються клінічні симптоми тяжкого прогресуючого захворювання з ураженням багатьох органів та систем. Відомі більше 40 лізосомних захворювань (ЛХ). Частота патології 1:5000. Щорічно в США народжується близько 200000 дітей з ЛХ. В Україні частота цієї патології невідома, що пов’язано з недоліками в діагностиці. Причиною порушення активності ферментів при ЛХ є генетичний дефект. Більшість генів при ЛХ уже ідентифіковані і відома їх локалізація. Лізосомні хвороби характеризуються такими ознаками: 1) супроводжуються накопиченням тих чи інших сполук; 2) накопичувані сполуки завжди локалізуються в лізосомах; 3) сполуки можуть бути гомогенні та гетерогенні; 4) дефіцит лише за одним фактором. Тип спадкування автосомно-рецесивний (АР), рідше рецесивний, зчеплений з Х-хромосомою. Класифікація: виділяють мукополісахаридози (синдроми: Гурлера, Шейє, Пантера, Сан-Філіппо, Моркіо, Мор ото-Ламі, Слав), сфігмоліпідози (синдроми: Тея-Сакса, Німана-Піка, Крабе, Фарбера, Шіндлера, GM 1, GM 2, метахроматична лейкодистрофія), муколіпідози (глікопротеїнози хвороба Вольмана, цероїдний ліпофусциноз, муколіпідози 1-3-го типів). Запідозрити патологію можна на основі клініки: для хворих характерна зовнішність з грубими рисами обличчя, наявні різні неврологічні порушення, скелетні аномалії, патологія зору та слуху, різні ступені розумової відсталості. Маніфестація захворювання може спостерігатися від періоду новонародженості до зрілого віку. Вирізняють ранні та пізні форми захворювання. Для виявлення використовують біохімічну діагностику (виявлення специфічних метаболітів у тканинах, наприклад глікозаміногліканів), знаходження генетичних дефектів у лейкоцитах та фібробластах шкіри хворого. Для пренатальної діагностики використовують культуру клітин амніотичної рідини, яку одержують при амніоцентезі. Мукополісахаридози – хвороби накопичення. В результаті нестачі лізосомальних ферментів змінюється катаболізм глікозаміногліканів та настає їх накопичення в лізосомах, що викликає грубу клітинну патологію та характерну клінічну картину. Захворювання успадковувається за АР-типом, за винятком синдрому Гантера (рецесивний, зчеплений з Х-хромосомою). За наявним ферментативним дефектом вирізняють 14 типів та підтипів мукополісахаридозів. Зазвичай захворювання проявляються у віці 1-6 місяців. Виникають і поступово розвиваються розумова відсталість, фізична неповноцінність, які часто призводять до смерті. Основні симптоми захворювання: затримка психічного розвитку, неврологічні порушення, підвищеність сухожилкових рефлексів, судоми, атаксія, дефекти зору (помутніння рогівки, катаракта та ін.), порушення слуху, кісткові деформації, вади серця, гепатоспленомегалія. Клінічна картина захворювань досить різноманітна і включає ураження сполучної тканини, які в одних випадках більш виражені в скелеті чи його частинах, в інших – в ураженні серця чи рогової оболонки очей. Біохімічним маркером хвороб накопичення є гіперсекреція і/чи внутрішньоклітинне накопичення глікозаміногліканів. Нині існує перспектива лікування мукополісахаридозів рекомбінантними препаратами (для лікування мукополісахарідозів 1-го та 4-го типів, хвороби Фабрі. Сподіваємося, що ці препарати в найближчому майбутньому з’являться і в Україні. Для синдрому Гурлера характерні раннє помутніння рогової оболонки, карликовість, гіпертрихоз, скіфоцефалія, макроцефалія, розумова відсталість, грубі риси обличчя, втрата слуху, гепатоспленомегалія, кили (грижі), викривлення колінних суглобів, брахідактилія, кігтеподібна кисть, розширення діафізів. Для синдрому Хантера характерні дизостози з карликовістю, розумова відсталість, гепатоспленомегалія, кардіопатія, відсутність помутніння рогівки, атиповий пігментний ретиніт, набряк диска зорового нерва, збільшене турецьке сідло, дзьобоподібні тіла поперекових хребців. Для синдрому Санфіліппо (А, В, С) характерні помірна карликовість, гіпертрихоз, середньої важкості ураження скелета, ущільнення кісток склепіння черепа, помірне огрубіння рис обличчя, відсутність помутніння рогівки, зниження слуху, незначна гепатомегалія, двоопуклі тіла поперекових хребців, розумова відсталість. Проявами синдрому Моркіо є карликовість, ущільнення кісток склепіння черепа, помутніння рогівки, втрата слуху, помірна гепатомегалія, гіпоплазія зубоподібного відростка, підвивих шийних хребців, цервікальна мієлопатія, кільоподібна деформація груднини, загальна слабкість м'язів, дисплазія стегон, збережений інтелект. Синдром Шейє клініка така сама, як і при синдромі Гурлера, але наявні тугорухливість суглобів, аортальна регургітація, але інтелект нормальний. Деякі дослідники вважають його синдромом Гурлера. Синдром Марото-Ламі – симптоматика як при синдромі Гурлера, але інтелект нормальний. Синдром Слая – грубі риси обличчя, різний ступінь помутніння рогівки, зниження слуху, гепатомегалія та спленомегалія, деформація грудної клітки, Х-подібна деформація ніг, збережений інтелект. Синдром ді Ферранте — низькорослість, розумова відсталість, гірсутизм, грубе волосся, гепатомегалія, помірний множинний дизостоз, гіпоплазія зубоподібного відростка. 9-й тип - низькорослість, множинні відкладення гіалуронової кислоти у навколосуглобових тканинах.
Дата добавления: 2015-12-16 | Просмотры: 680 | Нарушение авторских прав |