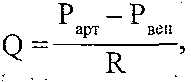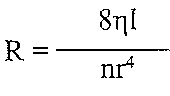|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
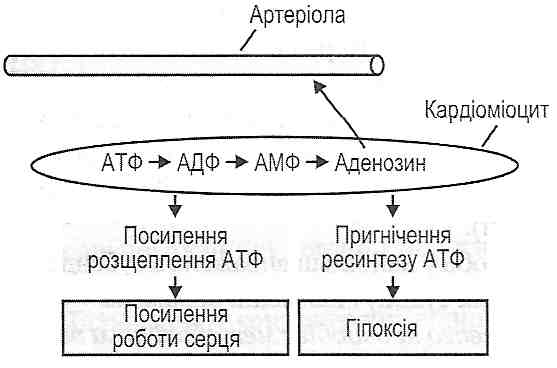
Залізодефіцитні анемії у дітей: причини і механізми розвитку, типові зміни периферичної крові, патогенез основних клінічних проявів. Залізорефрактерні анемії. 5 страницаАритмогенну кардіоміопатія або, користуючись прийнятої в медицині термінологією, аритмогенна правошлуночкова кардіоміопатія, відрізняється неухильним заміщенням м'язової серцевої тканини правого шлуночка серця на жирову і фіброзну (сполучну) тканини. Тобто, порушується скорочувальна функція серцевого м'яза за рахунок «сторонніх» серцю тканин, не здатних скорочуватися. Сучасна медицина схильна вважати, що аритмогенного кардіоміопатія має спадковий характер виникнення. Аритмогенну кардіоміопатія може існувати в організмі поряд з міокардитом, який буде вторинним і виникати внаслідок вразливості серцевого м'яза вірусів і інфекцій. Аритмогенну кардіоміопатія зустрічається у людей різних віків, аж до глибокої старості. У ряді випадків аритмогенного кардіоміопатія діагностується у спортсменів. Симптоми аритмогенну кардіоміопатії, як правило, починають проявлятися до досягнення пацієнтом 40-річного віку і виражаються в аритмії, тахікардії, запамороченнях, симптомах серцевої недостатності. 113. Недостатність вінцевого кровообігу: визначення поняття, причини і механізми розвитку, клінічні прояви. Механізми ішемічного і реперфузійного пошкодження кардіоміоцитів. 1. Високий рівень екстракції кисню в капілярах серця. У серці екстрагується 70—75 % 02, що надходить з артеріальною кров'ю, тимчасом як у тканинах головного мозку — 25 %, у скелетних м'язах і печінці — близько 20 %, у нирках — 10 %. Високий рівень вилучення 02 у серці пояснюється значною довжиною його капілярів і у зв'язку з цим — більшим часом контакту крові зі стінкою капілярів. При збільшенні потреби серця в кисні вона не може бути задоволена збільшенням екстракції 02 (як у скелетних м'язах), оскільки остання і так є максимально можливою в стані спокою. Тому для забезпечення збільшених енергетичних потреб серця залишається тільки один шлях — збільшення вінцевого кровообігу. 2. Високий базальний тонус вінцевих судин. Він дає можливість у стані спокою забезпечувати вінцевий кровообіг на рівні 250-300 мл/хв, що становить близько 5 % хвилинного об'єму крові. Високий базальний тонус вінцевих судин обумовлює високий резерв вінцевого кровообігу. Так, при зменшенні базального тонусу судин серця Інтенсивність кровообігу у них може зростати в 7—10 разів. 3. Фазний характер вінцевого кровообігу, пов'язаний з періодами серцевого циклу. Під час систоли відбувається здавлювання інтрамуральних судин - кровообіг мінімальний і становить близько 15 % загального вінцевого кровообігу. Під час діастоли здавлювання судин припиняється і кровообіг стає максимальним (близько 85 % загальної величини). Фазність вінцевого кровообігу найбільш виражена в субендокардіальній зоні міокарда (найбільше здавлювання судин) і найменш виражена - у субепікардіальній зоні. 4. Підпорядкованість вінцевого кровообігу метаболічним потребам серця і відносна незалежність його від нервових регуляторних впливів. В умовах патології ця підпорядкованість часто порушується і збільшується чутливість вінцевих судин до нервових імпульсів. 5. Винятково висока чутливість вінцевих судин до зменшення напруги кисню в крові. Зменшення р02 артеріальної крові всього лише на 5 % істотно збільшує інтенсивність вінцевого кровообігу. 6. Недостатній розвиток колатеральних судин. При несприятливих умовах колате-ралі в серці не можуть компенсувати порушення течії крові у вінцевих судинах, тому колатеральний кровообіг тут функціонально неповноцінний. 27.46. Як здійснюється регуляція вінцевого кровообігу? У регуляції вінцевого кровообігу розрізняють міогенну ауторегуляцію, метаболічну і нервову регуляцію. 1. Міогенна ауторегуляція. її основу становить закон Бейліса, відповідно до якого при розтягненні гладких м 'язів кровоносних судин збільшується сила їх скорочення. Міогенна ауторегуляція забезпечує сталість вінцевого кровообігу і відносну незалежність його від змін артеріального тиску. Так, при збільшенні тиску крові в аорті збільшується розтягування гладком'язових клітин вінцевих артерій, що веде до їх скорочення, підвищення тонусу артерій і збереження сталості кровообігу. При зменшенні артеріального тиску вінцевий кровообіг підтримується на постійному рівні завдяки розслабленню гладких м'язів і розширенню артерій. Нині показано, що при розтягуванні гладком'язових клітин судин збільшується проникність їхньої плазматичної мембрани до іонів кальцію. Останні проникають у клітини і викликають їх скорочення. 2. Метаболічна регуляція. Підпорядковує вінцевий кровообіг метаболічним потребам серця. Здійснюється за допомогою цілого ряду іонів і метаболітів, серед яких іони водню, калію, молочна кислота, простагландини. Однак найбільше значення мають два фактори: зменшення напруги 02 в артеріальній крові і аденозин. Останній утворюється в результаті гідролізу аденінових нуклеотидів при гіпоксії і при посиленій роботі серця. Будучи природним блокатором Са-каналів, аденозин зменшує надходження іонів Са2+ у цитоплазму гладком'язових клітин вінцевих судин, унаслідок чого зменшується ступінь їхнього скорочення і падає базальний тонус - вінцеві судини розширюються, коронарний кровообіг зростає (рис. 129). Рис. 129. Один з механізмів метаболічної регуляції вінцевого кровообігу 3. Нервова регуляція. Нейрогенний тонус вінцевих судин незначний, про що свідчить майже повна відсутність змін вінцевого кровообігу після повної денерва-ції судин серця. Набагато більше значення має опосередкований вплив нервової системи на коронарний кровообіг. Він здійснюється через зміни роботи серця та інтенсивності обміну речовин у ньому. В експерименті показано можливість і безпосереднього впливу нервів на тонус вінцевих судин. Так, при подразненні парасимпатичних нервів і введенні ацетилхоліну відбувається незначне розширення вінцевих артерій. Медіатори симпатичної нервової системи (катехоламіни) при дії на а-адренорецептори викликають звуження судин, а впливаючи на р-адренорецептори — розширення. Оскільки в нормі у вінцевих судинах переважають β-адренорецептори, то загальний ефект симпатичних впливів — незначне розширення судин серця. 27.47. Що таке недостатність вінцевого кровообігу? Чим відрізняються відносна і абсолютна коронарна недостатність? Недостатність вінцевого кровообігу - це патологічний стан, що характеризується нездатністю вінцевих судин забезпечувати кровопостачання серця відповідно до його енергетичних потреб. Інакше кажучи, виникає невідповідність між енергетичними потребами серця і доставкою кисню і поживних речовин вінцевими судинами. Недостатність вінцевого кровообігу може бути відносною і абсолютною. Відносна коронарна недостатність виникає у випадку первинного збільшення енергетичних потреб серця (збільшення навантаження на серце при фізичній роботі, артеріальній гіпертензії). За таких обставин інтенсивність вінцевого кровообігу може зростати, але цього буває замало, щоб задовольнити потреби серця. Абсолютна коронарна недостатність виникає у випадку первинного порушення вінцевого кровообігу, у результаті чого зменшується доставка кисню і поживних речовин міокарду як у стані спокою, так і при збільшенні енергетичних потреб серця. 27.48. Які патогенетичні фактори можуть обумовлювати розвиток абсолютної недостатності вінцевого кровообігу? Інтенсивність вінцевого кровообігу визначається формулою
де Q — об'ємна швидкість течії крові; Ра і Рвсн — тиск крові відповідно на початку і в кінці системи вінцевих судин; (Ра - Р) - перфузійний тиск; R - опір вінцевих судин. Оскільки абсолютна коронарна недостатність характеризується зменшенням інтенсивності вінцевого кровообігу (зменшенням Q), можливими є два патогенетичних варіанти її розвитку. І. Зменшення перфузійного тиску. При цьому розвивається коронарна недостатність центрального походження. її причинами можуть бути: а) артеріальна гіпотензія (зменшення Ра п), наприклад, при всіх видах шоку, колапсі, недостатності аортальних клапанів. При зменшенні артеріального тиску спрацьовують механізми міогенної ауторегуляції вінцевого кровообігу, що підтримують його на постійному рівні. Однак, коли артеріальний тиск падає нижче 70 мм рт. ст., ці механізми виявляються неспроможними; б) порушення венозного відтоку (збільшення Рвен). В експерименті моделюють перев'язуванням вен серця. Може мати значення при декомпенсованій недостатності правого шлуночка, коли збільшується центральний венозний і кінцеводіастолічний тиск. II. Збільшення опору вінцевих судин. При цьому розвивається коронарна недостатність місцевого походження. Оскільки
де tj — в'язкість крові; 1 — довжина судин; г — радіус судин, то збільшення опору може бути обумовлене: а) збільшенням в 'язкості крові при порушенні її реологічних властивостей (зневоднення, поліцитемія, ДВЗ-синдром). При цьому розвиваються порушення мікроциркуляції, аж до сладж-синдрому та істинного капілярного стазу;: б) зменшенням радіуса судин. Це основний фактор розвитку абсолютної коронарної недостатності. Він викликає ішемію серця. 27.49. Які механізми можуть лежати в основі розвитку ішемії міокарда? I. Обтураційний механізм — зменшення просвіту вінцевих артерій. Його причинами можуть бути: а) стенозуючш атеросклероз (є причиною ішемії міокарда у 90 % випадків); б) тромбоз вінцевих артерій (найчастіше є наслідком атеросклерозу); в) емболія вінцевих артерій; г) запальні процеси в стінці судин серця — коронаріїти. Бувають при ревматизмі, сифілісі. Для розвитку клінічних ознак ішемії міокарда має значення величина "критичного стенозу ". Це мінімальне зменшення просвіту судин, при якому виникає ішемія. У людини цей показник становить 75 % (у свиней - менше, у собак - більше). II. Ангіоспастичний механізм- спазм вінцевих судин. Його причинами можуть бути: а) збудження а-адренорецепторів на тлі блокади р-адренорецепторів; б) вазопресин; в) ангіотензин II; г) тромбоксан А^; ґ) гіпокапнія; д) ендотелій - біологічно активна речовина ендотеліального походження. Існує клінічна форма коронарного ангіоспазму — стенокардія Принцметала. III. Компресійний механізм — здавлювання вінцевих судин. Може мати місце при тахікардії (збільшується загальна тривалість періоду здавлювання вінцевих судин під час систоли серця). Іноді його причиною є рубці і пухлини. В експерименті використовують для моделювання ішемії й інфаркту міокарда шляхом накладення лігатури на вінцеві артерії. 27.50. Які патогенетичні фактори впливають на міокард в умовах коронарної недостатності? 1. Гіпоксія. 2. Ацидоз. Розвивається в результаті накопичення кислих продуктів обміну речовин унаслідок порушення відтоку крові і активації гліколізу. 3. Збільшення позаклітинної концентрації іонів калію в зоні ішемії. Виявляють від самого початку порушень вінцевого кровообігу (через кілька хвилин), коли ще немає ушкодження кардіоміоцитів. У цей період з невідомих ще причин відбувається пасивний вихід іонів калію з клітин (можливо, збільшується проникність К-каналів), і його позаклітинна концентрація зростає до 8 ммоль/л і більше. Через декілька годин "витікання" іонів К+ із клітин може бути обумовлене порушенням роботи Na-K-насосів (дефіцит АТФ) та збільшенням проникності ушкоджених мембран.
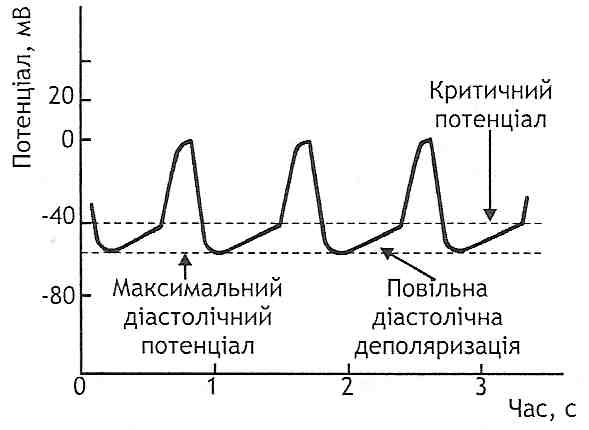
27.51. Які наслідки для міокарда може мати недостатність вінцевого кровообігу? 1. Порушення скорочувальної здатності міокарда з розвитком недостатності серця. 2. Поява аномальної електричної активності — електрична нестабільність серця, розвиток аритмій. 3. Ушкодження кардіоміоцитів, обумовлене ішемією (див. розд. 11). 4. Реперфузійний синдром. 27.52. Які порушення скорочувальної функції міокарда можуть виникати при його ішемії? При ішемії міокарда розрізняють ранні і пізні порушення його скорочувальної функції. Ранні порушення виникають дуже рано, вже при незначному дефіциті АТФ. Вони є відображенням гіпокальцієвого варіанта порушень скорочувальної функції серцевого м'яза і розвиваються в результаті блокади Са-каналів сарколеми кардіоміоцитів. Порушення провідності Са-каналів в умовах ішемії має щонайменше два механізми: а) дефіцит АТФ → порушення фосфорування білків Са-каналів; б) активація гліколізу →накопичення в клітинах іонів Н+ → безпосередня блокада Са-каналів. Наслідком зазначених порушень є зменшення надходження іонів Са2+ у саркоплазму м'язових волокон, розлади електромеханічного спряження, зменшення сили скорочень кардіоміоцитів. Пізні порушення скорочувальної функції виникають при тривалих розладах вінцевого кровообігу - понад ЗО хв. їх відносять до гіперкальцієвого (контрактурного) типу порушень. Концентрація іонів кальцію в саркоплазмі кардіоміоцитів збільшується через дві основні причини: а) порушення видалення іонів Са2+ із саркоплазми внаслідок дефіциту АТФ; б) збільшення надходження іонів Са2+ у м'язові волокна через ушкоджену плазматичну мембрану клітин. У результаті збільшення вмісту Са2+ порушуються процеси розслаблення кардіоміоцитів, настає контрактура міофібрил, порушується скорочувальна здатність серця. 114. Ішемічна хвороба серця: види, етіологія, патогенез, клінічні прояви. Патогенез проявів та ускладнень інфаркту міокарда. Ішемічна хвороба серця — це хвороба, що розвивається в результаті абсолютної недостатності вінцевого кровообігу й виявляється ушкодженнями міокарда різного ступеня тяжкості. її основними клінічними формами є: 1) стенокардія — напади короткочасної (до 20 хв.) гострої коронарної недостатності, які супроводжуються больовим синдромом, відчуттям страху і пов'язаними з цим вегетативними реакціями. Розрізняють стенокардію напруги, стенокардію спокою і стенокардію Принцме-тала (спазм вінцевих артерій); 2) передінфарктний стан (проміжний коронарний синдром, або гостра вогнищева дистрофія міокарда) — розвивається при тривалості'ішемії міокарда від 20 до 40 хв.; 3) інфаркт міокарда— некроз серцевого м'яза, обумовлений порушеннями вінцевого кровообігу. Виникає при оборотній (транзиторній) ішемії, що триває понад 40-60 хв.? або при необоротних порушеннях коронарного кровообігу; 4) кардіосклероз — склеротичні зміни серцевого м'яза. Можуть бути дифузними (атеросклеротичний кардіосклероз) і вогнищевими (постінфарктний кардіосклероз). 27.56. Назвіть основні причини розвитку інфаркту міокарда. 1. Атеросклероз вінцевих артерій (див. розд. 28). Його розвиток супроводжується порушенням постачання міокарда киснем. 2. Збільшення навантаоїсення на серце (фізична напруга, артеріальна гіпертензія). При цьому збільшується потреба серця в кисні. 3. Стрес (див. розд. 33). 27.57. Яка роль катехоламінів у розвитку інфаркту міокарда? У розвитку інфаркту міокарда можуть мати значення такі ефекти катехоламінів. 1. Порушення вінцевого кровообігу. Це пов'язане з тим, що катехоламіни: а) сприяють розвитку атеросклерозу (стрес і катехоламіни, що беруть участь у його реалізації, є фактором ризику цієї хвороби); б) викликають контрактурний спазм гладких м'язів вінцевих артерій (катехо-ламінове ушкодження клітин судинної стінки); в) активують тромбоутворення і зсідання крові. 2. Збільшення потреби серця в кисні. Енергетичні потреби серця зростають у результаті реалізації позитивного іно- і хронотропного ефектів катехоламінів та збільшення загального периферичного опору, внаслідок чого збільшується навантаження на серце. 3. Некоронарогенні катехоламіновіушкодження кардіоміоцитів. Вони не пов'язані з порушенням вінцевого кровообігу. Виникають у результаті того, що великі дози катехоламінів активують ліпідні і кальцієві механізми ушкодження клітин (див. розд. 11). 27.58. Які клінічні синдроми характерні для інфаркту міокарда? 1. Больовий синдром. 2. Гостра серцева недостатність. Розвивається при уражені великих ділянок міокарда. Може виявляти себе синдромом серцевої астми і набряку легень або карді-огенним шоком. 3. Аритмічний синдром. Можливий розвиток усіх видів аритмій. Найнебезпечнішою є поява фібриляції шлуночків. 4. Резорбційно-некротичний синдром. 27.59. Які механізми розвитку і значення больового синдрому при інфаркті міокарда? У розвитку больового синдрому при некрозі серцевого м'яза мають значення: а) хімічні фактори, що з'являються в тканинах при ушкодженні клітин. Серед них іони Н+, К+, простагландини, лізосомні ферменти; б) зміни скорочувальних властивостей ішемізованої ділянки міокарда, у результаті чого відбувається патологічне розтягування (пролабування) стінки серця при його скороченні. Це веде до подразнення механорецепторів серця і розвитку болю. Патогенетичне значення больового синдрому в розвитку інфаркту міокарда полягає в тому, що: 1) біль є потужним чинником ініціації стресу і активації симпатоадреналової системи. Великі дози катехоламінів, що вивільняються при цьому, сприяють ушкодженню міокарда (див. запит. 27.57); 2) сильний біль викликає спочатку збудження, а потім і перезбудження життєво важливих центрів головного мозку (дихального, серцево-судинного). Ця обставина є важливим чинником розвитку кардіогенного шоку 27.60. Що таке кардіотенний шок? У яких формах він може виявляти себе? Кардіогенний шок — це шок, що виникає в результаті різкого падіння нагнітальної (насосної) функції серця. Це - найнебезпечніше ускладнення інфаркту міокарда, що часто призводить до смерті. Розрізняють 4 форми кардіогенного шоку. 1. Рефлекторна форма (больовий шок). Основним механізмом її розвитку є тривалий біль, що викликає активацію симпатоадреналової системи, яка переходить у гальмування. Це призводить до депресії скорочувальної функції серця, брадикардії, зменшення тонусу периферичних судин і падіння артеріального тиску. 2. Гіпокінетична форма (істинний кардіогенний шок). Основним фактором її розвитку є різке зменшення скорочувальної функції серця в результаті ішемічного ушкодження кардіоміоцитів. Істинний кардіогенний шок розвивається, коли площа ураженого міокарда перевищує 40 %. 3. Дискінетична форма. Виникає в результаті асинергії (неузгодженості) скорочень міокарда. Причиною такої асинергії є грубі ушкодження серця - аневризми, розрив міжшлуночкової перегородки, відрив хорд клапанів. 4. Аритмічна форма. Є наслідком важких аритмій. 27.61. Який патогенез кардіогенного шоку? У патогенезі кардіогенного шоку розрізняють кілька етапів. I етап - первинне падіння артеріального тиску. Всі патогенетичні фактори кардіогенного шоку (рефлекторна депресія, збільшення площі ушкодженого міокарда, асинергія серцевих скорочень, аритмії) викликають зменшення серцевого ви-штовху. Це, за законами гемодинаміки, призводить до зменшення хвилинного об'єму серця і падіння артеріального тиску. II етап - компенсаторний спазм артеріол. Характеризується активацією сим-патоадреналової системи, надходженням у кров катехоламінів, вазопресину, глкжо-кортикоїдів, утворенням ангіотензину II. Вивільнення потужних судинозвужувальних факторів викликає генералізований спазм артеріол, у результаті чого збільшується загальний периферичний опір. Зазначена реакція є компенсаторною і спрямована на попередження подальшого падіння артеріального тиску. III етап - вторинне падіння артеріального тиску. Тривалий спазм артеріол у периферичних тканинах викликає порушення мікроциркуляції і гіпоксію. Наслідком кисневого голодування є: а) ацидоз, що викликає депресію скорочувальної функції міокарда; б) розширення артеріол, що виникає в результаті накопичення в тканинах метаболі-тів-вазодилататорів ("метаболічний симпатоліз "); в) надходження у кров із тканин так званих ішемічних токсинів. Серед них велике патогенетичне значення має фактор депресії міокарда, що вивільняється з підшлункової залози. Усі зазначені зміни, погіршуючи скорочувальну функцію серця й "знімаючи" компенсаторний спазм артеріол, викликають подальше падіння артеріального тиску. IV етап - термінальні зміни. У результаті істотного падіння артеріального тиску (нижче 40 мм рт. ст.): а) ще більше порушується коронарний кровообіг і збільшується ішемія міокарда -зменшення скорочувальної функції міокарда прогресує; б) розвивається гостра ниркова недостатність (повністю припиняється клубочко-ва фільтрація, виникають анурія, інтоксикація); в) порушується мозковий кровообіг, розвивається гіпоксія головного мозку, виникають розлади функції життєво важливих центрів. Сукупність зазначених змін призводить до смерті. 27.62. У чому сутність резорбційно-некротичного синдрому, що розвивається при інфаркті міокарда? Резорбційію-некротіїчний синдром при інфаркті міокарда є наслідком надходження в кров продуктів розпаду змертвілої тканини серця. Він виявляє себе такими ознаками: а) гарячкою (див. розд. 15); б) нейтрофільним лейкоцитозом; в) збільшенням швидкості осідання еритроцитів (ШОЕ); г) ферментемією — появою в крові ферментів, що надходять із ушкоджених кардіоміо-цитів (креатинкіназа, аспартатамінотрансфераза, лактатдегідрогеназа І типу та ін.); j) аутоімунним синдромом (синдромом Дреслера). Розвивається в результаті кон-формаційних змін білків міокарда. Виявляється запаленням серозних оболонок організму - полісерозитом (перикардитом, плевритом, перитонітом). 27.63. Що таке некоронарогенні некрози серця? Як їх моделюють в експерименті? Некоронарогеншіми називають некрози серця, що виникають не в результаті недостатності вінцевого кровообігу, а через інші причини. Існує кілька експериментальних моделей некрозу серцевого м'яза, причина виникнення якого не пов'язана з патологією вінцевих судин. Ці моделі певною мірою відбивають ситуацію, що спостерігається в природних умовах. 1. "Гіпоксичний некроз міокарда. Може бути відтворений за допомогою різних видів гіпоксії: гіпоксичної, гемічної. При цьому на тлі загальної недостатності кисню в організмі, що сама по собі веде до підвищення навантаження на систему кровообігу, розвивається некротичне ушкодження м'язових волокон серця. Розвитку некрозу сприяє фіксація тварини в незручній позі, наприклад, розтягування у верстаті, або додаткове навантаження — біг у тредбані. 2. Електролітно-стероїдна кардіопатія з некрозом. За спостереженнями Сельє, при введенні щурам значної кількості солей натрію разом з деякими аніонами (сульфатними, фосфатними) у серці з'являються осередки ушкодження дегенеративно-некротичного типу, що часто супроводжуються гіалінозом судин інших органів. Ці ушкодження стають більшими або виникають при введенні меншої кількості солей, якщо одночасно вводити деякі стероїдні гормони надниркових залоз. На цьому тлі легше розвиваються і мають тяжчий перебіг ушкодження серця, викликані іншими причинами. Так, введення навіть невеликих доз норадреналіну, похідних кальциферолу, гіпоксія, м'язова напруга або, навпаки, значне обмеження рухливості ведуть до розвитку великого некрозу міокарда. Солі калію і магнію при цьому мають захисну дію. 3. Імунні ушкодження серця. Можливі при введенні в організм експериментальної тварини гетерогенної сироватки, що містить антитіла проти білків серця тварини даного виду (кардіоцитотоксини). Доведено також, що в організмі за певних умов можуть виникати антитіла і сенсибілізовані лімфоцити, які діють на тканини власного серця і спричиняють його ушкодження. Цьому сприяє проникнення в кров денатурованих компонентів некротизованих кардіоміоцитів. В експерименті аналогічний процес можна викликати введенням тварині суспензії міокарда зі стимулятором імунологічної реакції (ад'ювантом Фрейнда). Серце може бути ушкоджене і циркулюючими імунними комплексами антиген-антитіло, а також при фіксації на його структурах цитофільних антитіл типу IgE з наступною їх реакцією з антигеном. 4. Нейрогенні ураження серця. Дистрофічні зміни і некроз міокарда можна відтворити гострим або хронічним подразненням шийно-грудного вузла симпатичного стовбура, блукаючого нерва, гіпоталамуса, мозкового стовбура або інших відділів головного мозку. Введення в кров великих доз адреналіну або норадреналіну також веде до ураження серця. В основі механізму нейрогенних ушкоджень лежить невідповідність між рівнями функції, метаболізму і кровопостачання. Подразнення серцевих симпатичних нервів супроводжується значним збільшенням споживання кисню міокардом. При цьому збільшення вінцевого кровообігу є недостатнім (відносна коронарна недостатність), а тому розвивається гіпоксія міокарда. При склерозуванні вінцевих артерій невідповідність інтенсивності кровообігу рівневі обміну речовин виявляється ще в більшій мірі, що може виявитися катастрофічним як для серця, так і для організму в цілому. 115. Аритмії серця. Експериментальне моделювання. Причини, механізми порушень автоматизму, збудливості, провідності, типові електрокардіографічні прояви. 27.17. Що таке аритмії серця? Як їх класифікують? Аритміями серця називають порушення частоти, ритму, узгодженості й послідовності його скорочень. Аритмія – порушення ЧСС, ритмічності скорочень, локалізації водія ритму чи провідності (Мурашко В.В., Електрокардіографія). Розвиток аритмій може бути пов'язаний з порушеннями основних функцій провідникової системи серця: автоматизму, збудливості і провідності. На цьому ґрунтується класифікація аритмій, відповідно до якої виділяють: I. Аритмії, обумовлені порушеннями автоматизму. II. Аритмії, пов'язані з порушеннями збудливості. III. Аритмії, обумовлені порушеннями провідності. IV. Аритмії, пов'язані з поєднаними порушеннями збудливості і провідності. 27.18. Які аритмії серця можуть виникати в результаті порушення функції автоматизму? Розрізняють дві групи аритмій, пов'язаних з порушенням автоматизму серця. I. Номотопні аритмії. Генерація імпульсів до скорочення, як і в нормі, відбувається в синусно-передсердному вузлі. До цієї групи відносять: а) синусну тахікардію - збільшення частоти серцевих скорочень; б) синусну брадикардію - зменшення частоти серцевих скорочень; в) синусну (дихальну) аритмію - зміну частоти серцевих скорочень у різні фази дихального циклу (прискорення при вдиху і уповільнення при видиху). II. Гетеротопні аритмії - синдром слабкості синусно-передсердного вузла. Генерація імпульсів до скорочення відбувається не в синусно-передсердному вузлі, а в інших структурах провідникової системи, що є водіями ритму II і III порядку. Синдром розвивається в результаті зменшення активності або припинення діяльності синусно-передсердного вузла при ушкодженні його клітин або первинних функціональних порушеннях. При цьому можуть розвиватися такі види патологічних ритмів серця: а) передсердний повільний ритм — водій ритму міститься в структурах лівого передсердя, частота серцевих скорочень менше 70/хв.; б) атріовентрикулярний ритм — джерелом імпульсів є водії ритму II порядку (верхня, середня або нижня частина атріовентрикулярного вузла), частота серцевих скорочень залежно від місця генерації імпульсів зменшується від 70 до 40/хв; в) ідіовентрикулярний шлуночковий ритм — генерація імпульсів відбувається у водіях ритму III порядку (пучок Гісса або його ніжки), частота скорочень серця менше 40/хв. 27.19. Які причини і механізми розвитку синусної тахі- і брадикардії? Синусні тахікардія і брадикардія належать до групи номотопних аритмій, пов'язаних з порушеннями функції автоматизму. Здатність до автоматичного утворення імпульсів залежить від клітин, розташованих у провідниковій системі серця (β-клітини), у яких відбувається спонтанна повільна деполяризація клітинної мембрани в період діастоли (рис. 122). У результаті, при досягненні певного критичного рівня, виникає потенціал дії. Частота генерації імпульсів залежить від трьох чинників: а) максимального діастолічного потенціалу цих клітин; б) рівня критичного потенціалу, після досягнення якого виникає потенціал дії; і в) швидкості діастолічної деполяризації.
Дата добавления: 2016-03-26 | Просмотры: 614 | Нарушение авторских прав |