|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Залізодефіцитні анемії у дітей: причини і механізми розвитку, типові зміни периферичної крові, патогенез основних клінічних проявів. Залізорефрактерні анемії. 7 страница27.38. Від чого залежить сила скорочень окремих кардіоміоцитів? На силу скорочень м'язових волокон серця впливають: 1) концентрація іонів Са2+у саркоплазмі. Залежність тут така: що вищий вміст Са2+ у саркоплазмі, то більше утворюється комплексів Са2+ з тропоніном С, то більше вивільняється центрів зв'язування (активних центрів) на актинових міофіламентах, то більше утворюється "містків" між актином і головками міозину, то більшою буде сила скорочення м'язового волокна. При зменшенні концентрації Са2+ у саркоплазмі — навпаки; 2) ступінь спорідненості тропоніну С до іонів кальцію. Іони водню й неорганічного фосфату, зв'язуючись з тропоніном С, унеможливлюють взаємодію цього білка з Са2+, у результаті чого сила скорочень кардіоміоцитів зменшується; 3) стан скорочувальних білків — актину і міозину. Велике значення має взаємне розташування актинових і міозинових міофіламентів. Воно лежить в основі залежності, що її описує закон Франка-Старлінга. При дуже сильному розтягненні м'язових волокон кількість утворюваних актоміозинових "містків" зменшується - зазначений закон не "спрацьовує", сила скорочень серця падає; 4) концентрація АТФ, енергія гідролізу якого забезпечує ковзання м'язових філамен-тів один відносно одного. 27.39. Що лежить в основі розслаблення кардіоміоцитів? Які механізми забезпечують видалення іонів кальцію із саркоплазми волокон міокарда? Що може бути причиною порушень розслаблення серцевого м'яза? Основний процес, що визначає розслаблення кардіоміоцитів, — це видалення іонів кальцію із саркоплазми, у результаті чого концентрація Са2+ у ній зменшується і стає нижче 10"7 моль/л. При цьому комплекси Са2+ з тропоніном С розпадаються, тро-поміозин зміщується по відношенню до актинових філаментів і закриває їхні активні центри — скорочення припиняється. Існує три механізми видалення іонів Са2 + із саркоплазми кардіоміоцитів: 1) Са-насоси плазматичної мембрани і саркоплазматичного ретикулуму. Видаляють Са2+ у позаклітинне середовище і цистерни саркоплазматичного ретикулуму. Складовою їх частиною є Са-АТФ-аза, яка для здійснення активного транспорту іонів Са2+ використовує енергію АТФ; 2) Na-Ca-обмінний механізм. Видаляє іони Са2+ у позаклітинне середовище. Є різновидом вторинного активного транспорту (антипорту). Використовує енергію градієнта концентрацій іонів натрію по обидва боки плазматичної мембрани, тому залежить від роботи Na-K-насоса, що створює цей градієнт; 3) Са-акумулятивна функція мітохондрій. Активується тільки при значному підвищенні вмісту іонів Са2+ у саркоплазмі, що найчастіше буває в умовах патології. Видалення Са2+ із саркоплазми в матрикс мітохондрій відбувається за рахунок енергії, що вивільняється в процесі транспорту електронів по дихальному ланцюгу. Використання цієї енергії на активний транспорт іонів Са2+ у мітохондрії є альтернативою окисному фосфоруванню. Основними причинами порушень розслаблення кардіоміоцитів є: а) дефіцит АТФ. При цьому порушується енергозабезпечення Са- і Na-K-насосів, а також не відбувається розщеплення актоміозинових "містків", що утворилися в процесі скорочення; б) порушення роботи Са-транспортних систем. Відомі спадково обумовлені дефекти білків Са-насосів, що призводять до розвитку кардіопатії. Порушення розслаблення кардіоміоцитів виявляють себе розвитком м'язових контрактур. 27.40. Яке значення має АТФ у забезпеченні функцій клітин міокарда? Чим можуть бути обумовлені порушення енергетичного обміну в серцевому м 'язі? Енергія гідролізу АТФ у функціонально активних кардіоміоцитах забезпечує: 1) механічну роботу скорочень міофібрил (ковзання міофіламентів один відносно одного); 2) осмотичну роботу — активний транспорт іонів Са2+, Na +, К+ проти градієнтів концентрацій (робота іонних насосів); 3) фосфорування білків Са-каналів, фосфоламбану (білка Са-насосів саркоплазматичного ретикулуму). Розлади енергозабезпечення кардіоміоцитів можуть бути пов'язані з порушеннями: а) ресинтезу АТФ (гіпоксія, голодування, дефіцит вітамінів, зменшення активності ферментів енергетичного обміну); б) транспорту АТФ із мітохондрій до місць його використання (порушення креатин-кіназної транспортної системи); в) утилізації АТФ (зменшення АТФ-азної активності структур кардіоміоцитів). 27.41. Дайте порівняльну характеристику гіпо- і гіперкальцієвого варіантів недостатності серця. Сучасний рівень знань про молекулярні механізми скорочувальної функції серця дозволяє виділити два принципово різних патогенетичних варіанти недостатності серця: гіпокальцієвий і гіперкальцієвий, для яких характерне відповідно зменшення і збільшення концентрації іонів Са2+ у саркоплазмі кардіоміоцитів. Гіпокальцієвий варіант розвивається в результаті порушень збудження і електромеханічного спряження у волокнах міокарда. Це буває при аритміях (брадикардії різного походження, блокади), короткочасній ішемії міокарда (порушується фосфорування Са-каналів у результаті дефіциту АТФ), ацидозі (блокада Са-каналів іонами водню), гіпокальціємії. Виявляє себе зменшенням сили серцевих скорочень. Основний принцип лікування — підвищення вмісту іонів Са2+ у саркоплазмі кардіоміоцитів під час систоли серця. Для цього використовують: а) серцеві глікозиди; б) катехола-міни і р-адреноміметики; в) парні електричні стимули. Гіперкальцієвий варіант розвивається в результаті посиленого надходження іонів Са2+ у саркоплазму кардіоміоцитів з позаклітинного середовища (усі види ушкодження сарколеми, при яких підвищується її проникність) або внаслідок порушень видалення Са2+ із саркоплазми (дефіцит АТФ, порушення функції Са-транспортних систем). Виявляється розвитком контрактури (перескорочення) міофібрил, у результаті чого стає неможливим розслаблення м'язових волокон, а отже, і наступне їх скорочення. Основний принцип лікування — зменшення вмісту іонів Са2+ у саркоплазмі кардіоміоцитів. Для цього використовують: а) β-адреноблокатори; б) блокатори Са-каналів. 116. Позаміокардіальна недостатність серця. Ураження перикарда. Гостра тампонада серця, прояви та наслідки.
Позаміокардіальна недостатність розвивається в тих випадках, коли до серця притікає мало крові по венах або коли воно не в змозі прийняти всю кров, що надходить. Перше буває при гіповолемії (крововтрата) або різкому розширенні судин (колапс), друге - при накопиченні рідини в порожнині перикарда, що утруднює розширення порожнин під час діастоли. Накопичення рідини в порожнині перикарда може відбуватися швидко і повільно. Швидке накопичення відбувається внаслідок крововиливу при пораненні або розриві серця, при перикардиті, що швидко розвивається. Через погану розтяжність перикарда в порожнині підвищується тиск, що перешкоджає діастолічному розширенню сер- ця, виникає гостра тампонада серця. В експерименті цей процес моделюють введенням рідини в порожнину перикарда (О. Фохт). При цьому спостерігають, як зменшується кро-вонаповнення порожнин серця, знижується ударний об'єм і падає артеріальний тиск. Між внутрішньоперикардіальним і артеріальним тиском існує чітка обернена залежність: що більший внутрішньоперикардіальний тиск, то нижчий артеріальний. Венозний тиск при цьому підвищується. Вмикання компенсаторних механізмів при перикардиті відбувається рефлекторно за участю сигналів, що надходять з трьох рецепторних полів: 1) отворів порожнистих і легеневих вен — підвищення тиску на шляхах припливу; 2) аорти і сонних синусів (синокаротидні зони) — зниження тиску на шляхах відтоку; 3) перикарда — підвищення внутрішньоперикардіального тиску. При перетинанні блукаючих і депресорних нервів, а також при вимиканні рецепторних полів за допомогою новокаїну пристосувальні реакції не здійснюються - порушення кровообігу мають набагато тяжчий перебіг. При тампонаді серця мобілізація потужних механізмів компенсації, які ведуть до посилення скорочень серця (гомео- і гетерометричні механізми, інотропний ефект катехоламінів), малоефективна або неможлива. Тому діють тільки відносно малопотужні і енергетично невигідні механізми компенсації і підтримки артеріального тиску - збільшення частоти серцевих скорочень і звуження периферичних судин. Цим і пояснюється важкий клінічний перебіг гострої тампонади серця. При повільнішому накопиченні рідини в перикарді робота компенсаторних механізмів виявляється ефективнішою, підвищення внутрішньоперикардіального тиску протягом деякого часу може компенсуватися. Повільне накопичення рідини, що спостерігається при хронічному ексудативному перикардиті і гідроперикарді, супроводжується поступовим розтягуванням перикарда і збільшенням об'єму навколосерцевої сумки. Унаслідок цього внутрішньоперикардіальний тиск змінюється порівняно мало, а порушення кровообігу не виникають тривалий час
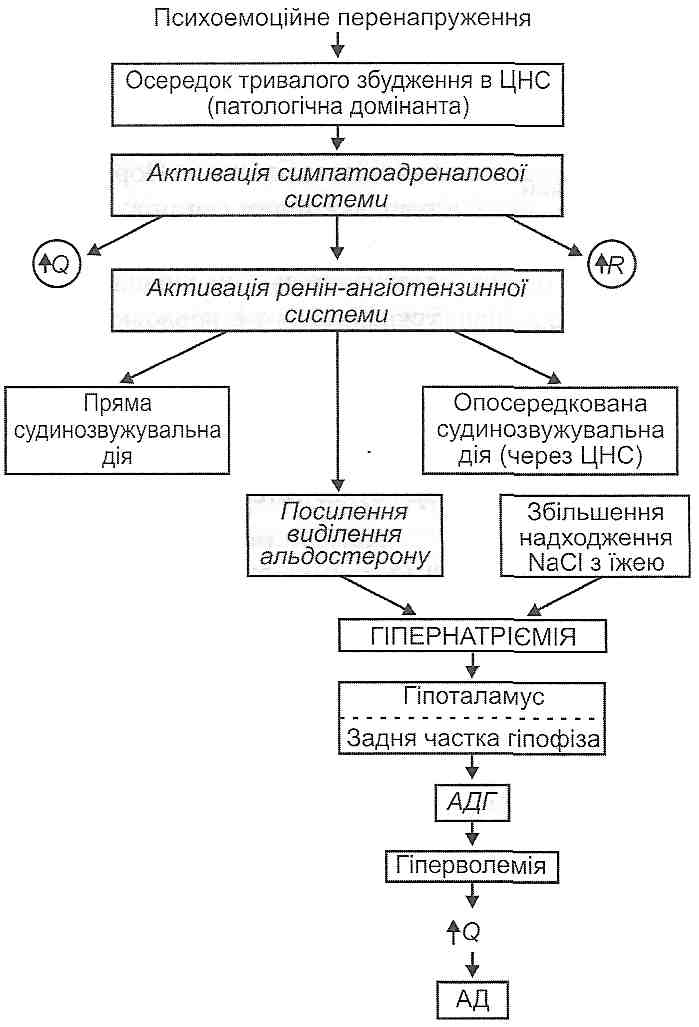
117. Артеріальна гіпертензія: визначення поняття, принципи класифікації. Первинна та вторинна артеріальна гіпертензія. Гемодинамічні варіанти. Особливості у дітей. 28.22. Що таке первинна і вторинна артеріальна гіпертензія? Розрізняють первинну і вторинну артеріальну гіпертензію. При первинній артеріальній гіпертензії підвищення артеріального тиску не може бути пов'язане з конкретним захворюванням або патологічним процесом у тих чи тих органах і системах: причина підвищення артеріального тиску залишається неясною. Для позначення цієї форми гіпертензії в різних країнах використовують два рівнозначних терміни: "есенціальна гіпертонія " і "гіпертонічна хвороба ". Вторинна артеріальна гіпертензія виникає як наслідок патологічних процесів у різних органах і системах. Вона характерна для: а) захворювань нирок (гломерулонефрит, пієлонефрит, полікістоз нирок та ін.); б) пухлин надниркових залоз (феохромоцитома, альдостерома); в) уражень серця й судин (деякі вади серця, коарктація аорти); г) захворювань нервової системи (бульбарний поліомієліт, енцефаліти; травми, струси мозку та ін.). У всіх цих випадках причина гіпертензії ясна. її усунення, як правило, веде до нормалізації артеріального тиску. 28.23. Які виділяють гемодинамічні варіанти артеріальної гіпертензії? Оскільки величина артеріального тиску визначається загальним законом гемоди-наміки, відповідно до якого P = QR, де Р — артеріальний тиск; Q — хвилинний об'єм серця; R — загальний периферичний опір, то артеріальна гіпертензія може бути обумовлена збільшенням хвилинного об'єму серця (Q), збільшенням загального периферичного опору (R) або тим та іншим одночасно. Відповідно до цього виділяють три гемодинамічних варіанти артеріальної гіпертензії. 1. Гіперкінетичнш тип. Обумовлений істотним збільшенням роботи серця, у результаті чого зростає його хвилинний об'єм (Q). 2. Еукінетичний тип. Виникає при помірному збільшенні хвилинного об'єму серця (Q) і загального периферичного опору (R). 3. Гіпокінетичний тип. Його розвиток пов'язаний з істотним збільшенням загального периферичного опору (R). 28.24. Які існують експериментальні моделі артеріальної гіпертензії? Жодна хвороба людини не має такої великої кількості різних експериментальних моделей, як артеріальна гіпертензія. Нині артеріальну гіпертензію вивчають на мишах, щурах, кролях, кішках, собаках, свинях, мавпах. За методами відтворення всі моделі артеріальної гіпертензії можна поділити на кілька великих груп. I. Порушення функції центральної нервової системи: а) зіткнення процесів умовного збудження і гальмування, що призводить до розвитку у тварин (собак, мавп) неврозу; б) моделювання психоемоційної напруги шляхом створення зоосоціального конфлікту (у мавп), змін біоритмів, іммобілізації тварин; в) електрична і хімічна стимуляція лімбічних структур головного мозку. II. Порушення мозкового крово- і лімфообігу: а) одно- і двостороннє перев'язування сонних і вертебральних артерій, що живлять мозок (центрально-ішемічна артеріальна гіпертензія)', б) блокада лімфовідведення по периневральних і периваскулярних лімфатичних шляхах за допомогою каоліну, що його вводять у велику цистерну мозку. III. Порушення функції депресорних регуляторних систем: а) двостороннє перетинання у кролів і собак депресорних і синусних нервів, у результаті чого знімаються гальмівні впливи з барорецепторів рефлексогенних зон дуги аорти і каротидного синуса (рефлексогенна гіпертензія, або гіпертензія розгальмування); б) центральна деаферентація барорецепторів, що її викликають ушкодженням ядра солітарного тракту; в) пригнічення синтезу простагландинів за допомогою індометацину. IV. Порушення функції нирок: а) звужування обох ниркових артерій або звужування однієї ниркової артерії з видаленням другої контрлатеральної нирки (реноваскулярна гіпертензія). Виникнення артеріальної гіпертензії в цьому випадку пов'язане з активацією ренін-ангіотензинної системи; б) видалення обох нирок і переведення тварин на гемодіаліз для запобігання уремії (ренопривна гіпертензія). її розвиток пояснюють припиненням депресорних функцій нирок; в) обгортання нирок целофаном, шовком. При цьому виникає перинефрит:' здавлюється ниркова паренхіма, розвивається венозний застій і гіпоксія нирок, активується ренін-ангіотензинна система. V. Порушення гормонального стану: а) введення тваринам адреналіну; в) введення вазопресину; в) субтотальне видалення кори надниркових залоз. При цьому відбувається посилення регенерації залозистої тканини з посиленою продукцією кортикостероїдів, особливо альдостерону (надшрнико-регенераційна гіпертензія). VI. Порушення водно-сольового обміну: а) введення тваринам великої кількості кухонної солі (сольова гіпертензія)', б) введення мінералокортикоїдів (дезоксикортикостерону, альдостерону) - мі-нералокортикоїдна гіпертензія; в) поєднане введення кухонної солі й мінералокортикоїдів. VII. Моделі генетично обумовленої артеріальної гіпертензії. У багатьох лабораторіях світу виведено чисті лінії щурів, характерною рисою яких є гіпертензія -ознака, що спадкується. Це, зокрема, щури зі спонтанною гіпертензією (лінія Окамото - Аокі); щури, схильні до інсультів; новозеландські щури, міланські щури; щури, чутливі до сольової дієти, та ін. 28.25. Яка етіологія первинної артеріальної гіпертензії? Нині виділяють ряд факторів, що мають безпосередній стосунок до виникнення гіпертонічної хвороби (есенціальної гіпертензії). 1. Психоемоційна перенапруга. Установлено більшу поширеність артеріальної гіпертензії серед людей, характер роботи яких пов'язаний з постійною психоемоційною напругою, наприклад серед телефоністок і телеграфістів, студентів у період екзаменаційної сесії, у дітей і підлітків, що навчаються у спеціалізованих математичних і деяких інших школах з напруженим режимом занять. Відомо, що під час другої світової війни в Ленінграді під час облоги виникла була ціла "гіпертонічна" епідемія. Російський учений Ланг уперше висловив думку, що першопричиною гіпертонічної хвороби є психоемоційні перенапруги, які ведуть до розвитку невротичних порушень вищої нервової діяльності і підвищення артеріального тиску як вегетативного компонента цих порушень. Основну роль Ланг відводив так званим "не-відреагованим" негативним емоціям, тобто таким, при яких сильні вегетативні реакції, зокрема з боку серцево-судинної системи, не супроводжуються адекватними руховими реакціями. 2. Спадковий фактор. Як доказ значення спадковості в етіології гіпертонічної хвороби наводять той факт, що артеріальну гіпертензію виявляють у парах однояйце-вих близнюків набагато частіше, ніж у парах близнюків двох'яйцевих. Крім того, поширеність артеріальної гіпертензії серед родичів хворих на гіпертонічну хворобу є значно більшою, ніж в популяції в цілому. Нарешті, експериментальні моделі генетично обумовленої гіпертензії у тварин за основними своїми характеристиками є найближчими до первинної артеріальної гіпертензії людини. 3. Надмірне споживання кухонної солі. В епідеміологічних дослідженнях відзначено низьку захворюваність на гіпертонічну хворобу в представників деяких етнічних груп, що живуть у різних регіонах нашої планети: гренландських ескімосів, аборигенів гір Китаю та Австралії, деяких племен індіанців, що живуть у Панамі. Загальним для всіх цих народностей є споживання малих кількостей кухонної солі (1—2 г на добу), якщо порівнювати з іншими людьми, що споживають її 10-1'5 г на добу. Поряд з цим, там, де солі споживається більше, первинна артеріальна гіпертензія виявляється частіше. Високу захворюваність на гіпертонічну хворобу виявлено серед негритянського населення Багамських островів, у деяких регіонах Японії, де споживання солі досягає 20-50 г на добу. В експерименті артеріальну гіпертензію можна одержати шляхом згодовування тваринам кухонної солі. 28.26. Які існують концепції патогенезу первинної артеріальної гіпертензії? Нині уявлення про патогенез гіпертонічної хвороби розвиваються в основному в рамках двох концепцій: дисрегуляторної і мембранної. Дисрегуляшорна концепція пояснює виникнення первинної артеріальної гіпертензії порушеннями механізмів регуляції артеріального тиску. В основі мембранної концепції лежить положення про те, що первинна артеріальна гіпертензія виникає як наслідок первинних порушень у гладком'язових клітинах артеріол. 28.27. Який патогенез первинної артеріальної гіпертензії з погляду дисрегуляторної концепції її розвитку? У патогенезі гіпертонічної хвороби розрізняють дві фази: гіперкінетичну і гіпо-кінетичену. Гіперкінетична фаза характеризується переважно збільшенням хвилинного об'єму серця, наслідком чого і є підвищення артеріального тиску (рис. 132). У її розвитку виділяють ряд послідовних етапів. I етап — активація симпатоадреналової системи. Відбувається в результаті частих стресів, психоемоційних перенапруг, що зумовлюють появу осередків постійного тривалого збудження в центральній нервовій системі (патологічна домінанта). Катехоламіни, які виділяються при цьому, викликають щонайменше три важливих для подальшого розвитку гіпертензії ефекти: а) збільшують хвилинний об'єм серця; б) збільшують загальний периферичний опір; в) викликаючи спазм приносних артеріол нирок і безпосередньо діючи на клітини юкстагломерулярного апарату, сприяють виділенню в кров реніну. II етап — активація ренін-ангіотензинної системи. Надходження реніну в кров зумовлює ряд послідовних біохімічних реакцій, у результаті яких утворюється ангіо-тензин Пі ангіотензин III. З цими пептидами пов'язані такі зміни: а) скорочення гладких м'язів артеріол (ангіоспазм); б) збудження структур центральної нервової системи, що беруть участь у регуляції артеріального тиску; в) вивільнення в кров альдостерону клітинами клубочкової зони кори надниркових залоз. III етап — активація альдостерон-вазопресинової системи. Надходження в кров альдостерону, а також надмірне надходження в організм хлориду натрію викликають розвиток гіпернатрієміїІ підвищення у зв'язку з цим осмотичного тиску плазми крові. Збудження центральних і периферичних осморецепторів, що настає в цих умовах, активує секрецію вазопресину (антидіуретичного гормону) у ядрах гіпоталамуса. Вазопресин, впливаючи на нирки, викликає збільшення факультативної реабсорбції води.Це, у свою чергу, веде до збільшення об'єму циркулюючої крові (гіперволемія), хвилинного об'єму серця, а отже, і артеріального тиску. Зазначений механізм доповнюється безпосередньою судинозвужувальною дією вазопресину.
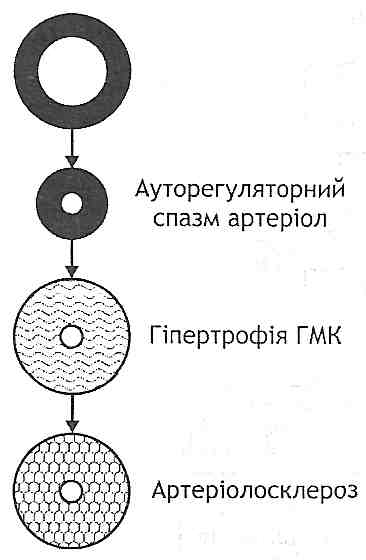
Рис. 132. Механізми гіпєркінетичноїфази розвитку первинної артеріальної гіпертензії: Q - хвилинний об'єм серця; R - загальний периферичний опір; AT— артеріальний тиск; АДГ - антидіуретичшй гормон Гіпокінетична фаза характеризується необоротними структурними змінами ре-зистивних судин, у результаті чого загальний периферичний опір і артеріальний тиск постійно збільшені (рис. 133). У розвитку цієї фази можна виділити ряд послідовних стадій: 1) ауторегуляторний спазм артеріол. Виникає як наслідок збільшення хвилинного об'єму серця. Є реакцією, спрямованою на підтримку сталості кровообігу в тканинах (попереджає надходження надлишкової кількості крові); 2) гіпертрофія гладких м 'язів артеріол. Є структурним проявом гіперфункції гладких м'язів, що виникає при часто повторюваних спазмах; 3) артеріолосклероз. Гіпертрофовані гладком'язові клітини поступово зазнають дистрофічних змін і гинуть, відбувається їх заміщення сполучною тканиною - розви- вається артеріолосклероз. Артеріоли перетворюються у ригідні сполучнотканинні трубки, не здатні ні до скорочення, ні до розслаблення. Загальний периферичний опір, а отже, і артеріальний тиск постійно збільшені. Порушується живлення життєво важливих органів: головного мозку, серця, нирок. Можливий розрив змінених артеріол — тоді розвивається крововилив. Найнебезпечнішим ускладненням є крововилив у мозок — геморагічний інсульт.
Рис. 133. Механізми гіпокінетичної фази розвитку первинної артеріальної гіпертензії 28.28. У чому сутність мембранної концепції патогенезу первинної артеріальної гіпертензії? Автори мембранної концепції (Ю. Постнов, Р. Орлов) головну роль у розвитку гіпертонічної хвороби відводять спадково обумовленим порушенням іонних насосів мембран гладком'язових клітин. У рамках цієї концепції нині розвиваються два напрями, що вивчають роль відповідно Са- і Na-K-насосів у порушенні функції гладком'язових клітин артеріол. 1. Дефекти Са-насосів клітинних мембран спричиняються до порушень видалення іонів кальцію із цитоплазми клітин і збільшення їхньої внутрішньоклітинної концентрації. Це викликає постійну контрактуру (перескорочення) гладких м'язів артеріол, що виявляється збільшенням загального периферичного опору і артеріального тиску. Крім того, надлишок іонів Са2+ у цитоплазмі клітин викликає їх ушкодження (див. розд. 11) і є, таким чином, передумовою розвитку артеріоло-склерозу. 2. Пригнічення роботи Na-K-насосів плазматичної мембрани гладком'язових клітин є однією з ознак первинної артеріальної гіпертензії. Про це, зокрема, свідчить той факт, що у багатьох хворих на гіпертонічну хворобу виявляють так званий ендогенний строфантиноподібний фактор, що, як і відомі серцеві глікозиди (строфантин, оуабаїн), пригнічує роботу Na-K-насосів. У результаті порушень діяльності Na-K-насосів у цитоплазмі поступово збільшується концентрація іонів Na+ і виникає набряк гладком'язових клітин артеріол. Це має кілька наслідків: а) потовщення стінки і зменшення просвіту артеріол; б) збільшення чутливості гладком'язових клітин до дії ендогенних катехоламінів; в) ушкодження і загибель клітин з наступним розвитком артеріосклерозу. Усі наведені зміни викликають стійке збільшення загального периферичного опору і підвищення артеріального тиску.
118. Причини і механізми розвитку вторинних артеріальних гіпертензій, експериментальне моделювання.
Дата добавления: 2016-03-26 | Просмотры: 585 | Нарушение авторских прав |