|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Патогенез ХСНЭтиологические факторы приводят к повреждению миокарда. В результате этого падает сократительная способность миокарда, что ведет к уменьшению сердечного, снижению УО и МОК. Все это приводит к тому, что органы и ткани организма не получают достаточного количества крови и кислорода, развивается ишемия их и гипоксия. В ответ на развитие ишемии и гипоксии включаются компенсаторные механизмы, которые на какой-то период времени обеспечивают нормальное функционирование ССС больного. Механизмы компенсации делят на: Кардиальные (закон Старлинга, гиперфункция, гипертрофия, ремоделирование) Экстракардиальные (активация нескольких нейрогуморальных систем)
В настоящее время общепринятой теорией патогенеза ХСН является нейрогормональная теория, согласно которой чрезмерная активация нейрогормональных систем в последующем приводит к гипертрофии миокарда, ремоделированию миокарда и сосудов, развитию гибернации миокарда, систолической и диастолической дисфункции левого желудочка. Современная нейрогуморальная модель патогенеза доказала, что развитие ХСН происходит по единым патофизиологическим законам вне зависимости от этиологии повреждения. С клинической точки зрения это дает «формальные» основания обозначить ХСН не только как сложный симптомокомплекс, осложняющий течение того и ли иного заболевания сердечно-сосудистой системы, но и как самостоятельную нозологическую форму. Нейрогормональные изменения при ХСН характеризуются следующим: · активацией симпатоадреналовой и снижением активности парасимпатической системы; · активацией ренин-ангиотензин-альдостероновой системы; · нарушением функционирования системы натриуретических пептидов; · дисфункцией эндотелия и дисбалансом между вазодилатирующими и вазоконстрикторными веществами; · повышением продукции вазопрессина (антидиуретического гормона); · гиперпродукцией определенных провоспалительных цитокинов (фактора некроза опухоли -ά); · повышением продукции вазоконстрикторных простогландинов; · активацией апоптоза кардиомиоцитов.
Гиперактивация симпатоадреналовой системы (САС) При снижении сердечного выброса у больных ХСН активируются барорецепторы синокаротидной зоны и дуги аорты, происходит активация симпатоадреналовой системы, при этом увеличивается концентрация адреналина и особенно норадреналина в крови. Активация симпатоадреналовой системы на начальном этапе ХСН оказывает определенное положительное адаптивно-компенсаторное влияние на сердечно-сосудистую систему. увеличение ЧСС и повышение сократительной способности миокарда вследствие стимуляции β1 –адренорецепторов миокарда, что приводит к увеличению МОК; развитие компенсаторной концентрической гипертрофии миокарда; стимуляция ά1 –адренорецепторов и повышение венозного тонуса, что приводит к увеличению венозного возврата крови к сердцу и увеличению преднагрузки; стимуляция ά1 –адренорецепторов артерий и артериол, что вызывает повышение общего сосудистого сопротивления; активация ренин-ангиотензиновой системы вследствие стимуляции β1 –адренорецепторов юкстогломерулярного аппарата почек
Указанные эффекты активации САС на адаптивно-компенсаторном этапе повышают сократительную способность миокарда, увеличивают венозный приток крови к сердцу (преднагрузку) и, следовательно, давление наполнения желудочков. Благодаря активации САС удается в течение определенного времени обеспечить достаточный сердечный выброс, МО, поддерживать на должном уровне АД и перфузию органов и тканей.
Однако продолжающаяся в течение длительного времени гиперактивация САС начинает оказывать отрицательное влияние на сердечно-сосудистую систему и способствует прогрессированию сердечной недостаточности вследствие: · чрезмерной констрикции вен и артериол (увеличение венозного притока – преднагрузки и росту периферического сопротивления – постнагрузки и снижению перфузии тканей); · увеличения объема циркулирующей крови (активация ренин-ангиотензин-альдостероновой системы и выраженная задержка натрия и воды); · значительного повышения потребности миокарда в кислороде; · развития тяжелых нарушений сердечного ритма; · непосредственного кардиотоксического эффекта (дистрофия миокарда, возможны даже некротические изменения); · развитие гибернации части кардиомиоцитов; · уменьшение плотности β –адренорецепторов (снижается чувствительность миокарда к катехоламинам); · повышенная агрегация тромбоцитов (что ухудшает кровоснабжение тканей и миокарда); · перегрузки кардиомиоцитов ионами кальция вследствие активации медленных кальциевых каналов с последующей пергрузкой кальцием митохондрий (наступает истощение запасов креатинфосфата и АТФ, разрушение кардиомиоцитов).
Гиперактивация ренин-ангиотензин-альдостероновой системы (РААС) В нормальных условиях 80% крови проходит через короткие нефроны, расположенные в корковом слое почек, а 15% через длинные расположенные в ЮГА. Ишемия их приводит к активации ЮГА и повышению выброса высоко активного фермента ренина. Под влиянием ренина активируется образование в печени из белка ангиотензиногена ангиотензина I (АТ I). АТ I является промежуточным продуктом и под влиянием АПФ расщепляется до ангиотензина II (АТ II), который является мощным вазоконстрикторным фактором. В активации РААС принимает участие также дисфункция сосудистого эндотелия. Это связано с тем, что основная часть АПФ расположена на мембране эндотелиальных клеток. При ХСН повышается активность эндотелиального АПФ, что сопровождается увеличением синтеза АТ II. АТ II с одной стороны повышает спазм сосудов, с другой – стимулирует продукцию альдостерона в клубочковой зоне коры надпочечников. Гиперсекреция альдостерона усиливает реабсорбцию натрия и воды в проксимальных и дистальных почечных канальцах, что приводит к увеличению ОЦК (ОЦК увеличивается также и за счет спазма сосудов – адреналин, норадреналин).
В настоящее время уставноленны особенности активирования миокардиальной ренин-ангиотензин-альдостероновой системы при ХСН. При ХСН на стадии декомпенсации активация кардиальной РААС происходит преимущественно в интерстициальном пространстве миокарда. На локальном (тканевом) уровне АТ II в основном синтезируется фибробластами, на долю кардиомиоцитов приходится всего лишь 10% рецепторов. В связи с чем важнейший компонент ремоделирования левого желудочка – периваскулярный фиброз коронарных артерий, обнаруживаемый на ранних стадиях ремоделирования.
Длительная гиперактивация РААС приводит к: · чрезмерное увеличение общего периферического сосудистого сопротивления (за счет спазма артериол), увеличение постнагрузки, снижение перфузии органов и тканей; · резко выраженная задержка натрия и воды (вследствие значительно увеличенной реабсорбции воды и натрия в почечных канальцах), значительное увеличение ОЦК, формирование отечного синдрома, увеличение преднагрузки; · повышение чувствительности миокарда к влияниям активированной САС и катехоламинам, в частности увеличение риска возникновения фатальных желудочковых аритмий; · потенцирование действия САС; · повышение потребности миокарда в кислороде под влиянием возрастающих постнагрузки и преднагрузки и продолжающейся активацией САС; · развитие гипертрофии, ремоделирования, апоптоза и фиброза миокарда с последующим снижением сократительной функции миокарда (гипертрофия миокарда и апоптоз кардиомиоцитов стимулируются АТ II, в развитии фиброза миокарда, вследствие стимуляции синтеза коллагена огромную роль играет гиперпродукция альдостерона, в процессах ремоделирования миокарда участвуют одновременно АТ II и альдостерон); · гипертрофия и ремоделирование сосудов с дальнейшим ростом ОПСС; · хроническая клубочковая гипертензия с последующим развитием в почках фиброза, гибелью клубочков почек, падением клубочковой фильтрации, развитием ХПН различной степени выраженности; · стимуляция секреции вазопрессина (антидиуретического гормона), который повышает реабсорбцию воды в почечных канальцах и увеличивает ОЦК и способствует развитию отечного синдрома (продукция вазопрессина ядрами гипоталамуса стимулируется АТ II); · ингибирование вазодилатирующей кининовой системы (АПФ обладает кининазной активностью). Необходимо отметить, что повышение секреции альдостерона происходит не только под влиянием АТ II, но и под воздействием вазопрессина, эндотелина, АКТГ. Синтезированный альдостерон попадает в кровь и связывается со специфическими рецепторами стромы и клубочков почек, слюнных желез, коры головного мозга, лейкоцитов, фиброблатов, эндотелиоцитов. Взаимодействие альдостерона с рецепторами фибробластов приводит к усилению синтеза коллагена, развитию фиброза, а взаимодействие с рецепторами эндотелиоцитов – к пролиферации эндотелия, гладкомышечных клеток, ремоделированию сосудов и миокарда.
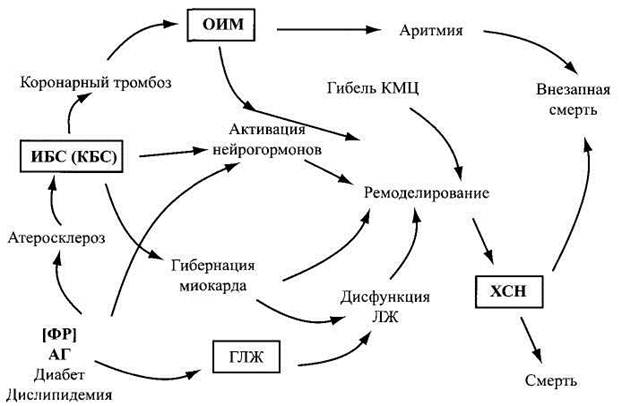
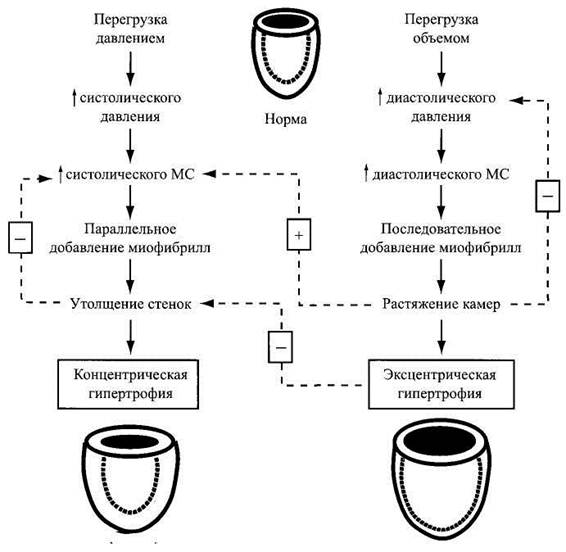
Рис. Сердечно-сосудистый континуум Рассмотрим два ключевых звена континуума. Первое из них — гипертрофия миокарда. Выделяют два типа гипертрофии. Концентрическая гипертрофия возникает при перегрузке давлением (артериальная гипертония, стенозы клапанных отверстий) и характеризуется повышением в основном конечного систолического давления в полостях, параллельным добавлением миофибрилл, утолщением стенки сердца при малорасширенных полостях. Эксцентрическая гипертрофия возникает при перегрузке сердца объемом (клапанные регургитации, врожденные аномалии с повышением конечного диастолического давления в полостях (в том числе при ишемическом или воспалительном поражении миокарда), последовательным добавлением миофибрилл, расширением полостей сердца при умеренном утолщении его стенок.
Рис. Развитие разных типов гипертрофии миокарда. Дата добавления: 2015-02-06 | Просмотры: 1409 | Нарушение авторских прав |