|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
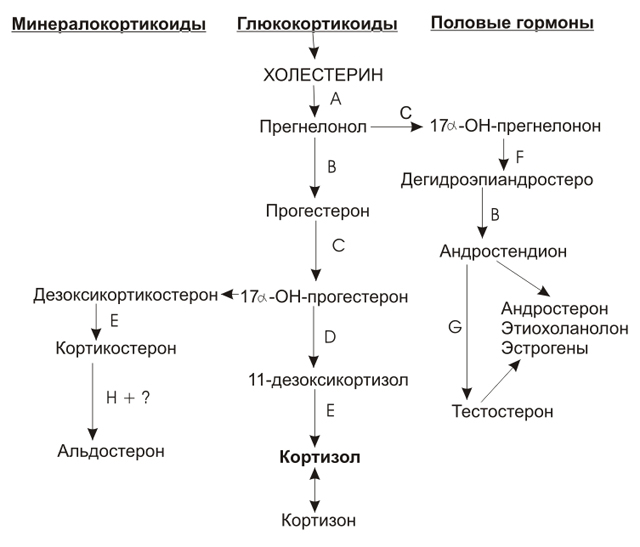
Первичная надпочечниковая недостаточностьТравма надпочечников. В периоде новорождённости чаще всего она является следствием родовой травмы надпочечников, как правило, при родах в тазовом предлежании. Это обусловлено и тем, что надпочечники имеют крупные размеры и интенсивное кровоснабжение. Кровоизлияние в надпочечники может произойти и при геморрагической болезни новорождённых, присоединении инфекционного заболевания. Если повреждена небольшая часть ткани надпочечников или повреждение было односторонним, то в периоде новорождённости они могут быть не диагностированы или случайно обнаружены в последующем в виде кальцификатов. У всех новорождённых с явлениями сердечно-сосудистого шока или коллапса, сочетающимися с гипонатриемией, необходимо исключить возможность развития надпочечниковой недостаточности с проведением УЗИ надпочечников и определением уровней кортизола и АКТГ в плазме или сыворотке крови. Дегенеративные изменения надпочечников. Наиболее частой причиной развития хронической надпочечниковой недостаточности является идиопатическая атрофия или дегенерация надпочечников, но в периоде новорождённости они выявляются крайне редко и при этом необходимо исключить врождённую гипоплазию надпочечников. При обследовании у новорождённых выявляется как глюко-, так и минералокортикоидная недостаточность. Семейная изолированная глюкокортикоидная недостаточность. Эта редкая врождённая патология может проявляться уже в периоде новорождённости сердечно-сосудистым шоком, гиперпигментацией и гипогликемией. В некоторых описанных случаях нельзя исключить и вариант АКТГ-резистентности. В литературе приводится описание семьи, в которой у 5 сиблингов отмечалась гиперпигментация, гипогликемия, судорожный синдром и лабораторно подтверждённая глюкокортикоидная недостаточность при сохранённой минералокортикоидной функции. При этом у 2-х сиблингов признаки надпочечниковой недостаточности в периоде новорождённости отсутствовали и появились в более старшем возрасте, что позволяло предположить врождённый характер выявленных изменений. Врождённая дисфункция коры надпочечников (врождённая гиперплазия надпочечников) – ВДКН. Этиология. Это наследственно обусловленное заболевание, в основе развития которого лежит дефект ферментов, участвующих в биосинтезе гормонов в коре надпочечников. Тип наследования аутосомно-рецессивный. Как видно из представленной схемы (рис. 4) основными биохимическими реакциями при биосинтезе из холестерина гормонов коры надпочечников являются процессы гидроксилирования, незаменимым коферментом которых является цитохром Р450. Для запуска процессов стероидогенеза необходимо, чтобы стероидогенный активный регуляторный протеин (СТАР) сформировал контактные участки между наружной и внутренней мембранами митохондрий, обеспечивая транспорт в них холестерина. При этом расщепление боковой цепочки холестерина опосредуется Р450sсс. Гидроксилирование как прогестерона, так и 17-ОН-прогестерона обеспечивается одним ферментом 21-гидроксилазой с участием Р450с (Р450с21). Также один и тот же кофермент, Р450с11, стимулирует активность 11β-гидроксилазы, 18-гидроксилазы и альдегидсинтетазы. Р450с17 является коферментом 17α-гидроксилазы и 17,20-лиазы. Некоторые варианты (изоферменты) 3β-гидроксистероиддегидрогеназы не требуют присутствия кофермента Р450. Ген, кодирующий один из таких ферментов, локализован на хромосоме 1, клонирован и доказано его участие в развитии ВДКН, обусловленном недостаточностью 3β-гидроксистероиддегидрогеназы.
Рис. 4. Схема биосинтеза стероидных гормонов в коре надпочечников. Обозначения: А – 20,22-десмолаза (Р450scc); В - 3β-гидроксистероиддегидрогеназа; С- 17α-гидроксилаза (Р450с17); D – 21-гидроксилаза (Р450с21);Е –11-гидроксилаза (Р450с11) F – 17,20-лиаза (Р450с17); G – 17-кеторедуктаза; Н – 18-гидроксилаза + 18-оксидаза (Р450с11).
Патогенез. В надпочечниках синтезируется 3 группы стероидных гормонов – минералокортикоиды, глюкокортикоиды и половые гормоны, в основном андрогены. Основным регулятором их биосинтеза является АКТГ, который оказывает также трофическое действие на железистую ткань надпочечников. При недостаточной продукции кортизола вследствие различных ферментативных дефектов по принципу обратной связи резко возрастает биосинтез гипофизом АКТГ. Оставаясь постоянно повышенным, АКТГ вызывает компенсаторную двустороннюю гиперплазию надпочечников, которая может частично компенсировать недостаточную продукцию гормонов. Вместе с тем, АКТГ резко увеличивает биосинтез и повышенную продукцию надпочечниками стероидов-предшественников основных гормонов. Различные варианты ВДКН представлены в табл. 7. Таблица 7. Клинико-лабораторные особенности различных форм ВДКН.
Вирильный синдром при ВДКН. Вирилизация наружных половых органов у плода женского пола при ВДКН происходит вследствие повышенной продукции надпочечниками андрогенов при недостаточности ферментов, которые принимают участие только в биосинтезе глюко- и минералокортикоидов. При постоянно повышенном уровне АКТГ и гиперстимуляции надпочечников в них происходит накопление промежуточных соединений, в частности 17-ОН-прогестерона, а активация вторичных путей их метаболизма (основные блокированы) усиливает образование андростендиона и тестостерона. Этому способствует и гиперплазия надпочечников. Как указывалось выше, гипоталамо-гипофизарно-надпочечниковая система начинает функционировать на 10-12 неделе беременности. С 10-й недели происходит также формирование наружных половых органов плода, а повышенное образование андрогенов в надпочечниках нарушает этот процесс. Происходит вирилизация или андрогенизация наружных половых органов плода. Классическими формами ВДКН с вирильным синдромом являются 21- и 11-гидроксилазная недостаточность. Синдром потери соли при ВДКН. Он развивается в результате недостаточной продукции альдостерона при дефекте ферментов 3β-гидроксистероиддегидрогеназы и полном блоке 21-гидроксилазы. При этом развивается гипонатриемия, гиперкалиемия и обезвоживание. Описан и вариант недостаточного биосинтеза альдостерона при дефекте 18-дегидрогеназы, последнего фермента по пути биосинтеза этого гормона, при этом другие проявления ВДКН, характерные для остальных его вариантов, отсутствуют. Синдром артериальной гипертензии. Он характерен для 11-гидроксилазной недостаточности, которая развивается вследствие дефекта кофермента Р450с11, в результате чего в надпочечниках происходит накопление дезоксикортикостерона, который хоть и является промежуточным соединением на пути биосинтеза альдостерона, уже обладает высокой минералокортикоидной активностью. Артериальная гипертензия отмечается и при недостаточности Р450с17 и дефекте фермента 17α-гидроксилазы и блокирования 17-гидроксилирования прогестерона. Синдром неполной маскулинизации. Развивается у мальчиков с вариантами ВДКН, которые обусловлены недостаточностью ферментов, участвующих в биосинтезе тестостерона. Формирование наружных половых органов у плода мужского пола происходит нормально только при условии достаточной продукции андрогенов тестикулами, что предполагает сочетанный ферментативный дефект и в них. Была продемонстрирована недостаточность 3β-гидроксистероиддегидрогеназы одновременно в надпочечниках и тестикулах, что предполагает общий генетический контроль биосинтеза тестостерона в этих железах внутренней секреции. Синдром липоидной гиперплазии. Этот синдром обусловлен двойным дефектом: 1) нарушением стероидогенеза вследствие генетической мутации СТАР-протеина; 2) повреждением клеток за счёт накопления в митохондриях эфиров холестерина. Вследствие полного блока биосинтеза андрогенов (по-видимому, как в надпочечниках, так и в ткани тестикул) наружные половые органы плода обоего пола под влиянием автономной тенденции к феминизации формируются по женскому типу. Классификация и клиническая картина. В настоящее время выделяют три основные формы ВДКН, которые составляют более 90% всех случаев болезни: Простая, вирильная или компенсированная форма. Она обусловлена частичной недостаточностью 21-гидроксилазы. Единственным клиническим признаком болезни при рождении является неправильное строение наружных половых органов. У мальчиков наружные половые органы сформированы в целом правильно, по мужскому типу, но с различной степенью выраженности макрогенитосомии (увеличения размеров) и пигментации. При незначительной выраженности этих изменений в периоде новорождённости их могут не заметить. У девочек наружные половые органы сформированы неправильно, с различной степенью выраженности вирилизацией. При лёгкой степени вирилизации отмечается только гипертрофия клитора, его форма не изменена и в ряде случаев эти изменения у новорождённых девочек не замечают. При более выраженной средней степени вирилизации (наблюдается наиболее часто) выявляется гермафродитное строение гениталий: клитор не только гипертрофирован, но и становится пенисообразным, формируется головка и крайняя плоть; уретра открывается одним отверстием с влагалищем, формируя sinus urogenitalis с узким воронкообразным входом во влагалище; малые половые губы гипоплазированы, большие половые губы становятся складчатыми и пигментированными. В более редких случаях при значительной вирилизации большие половые губы могут срастаться, напоминая расщеплённую мошонку; клитор с пенильной уретрой; может отмечаться атрезия наружных двух третей влагалища. Тяжёлая, сольтеряющая форма. Эта форма обусловлена полным блоком 21-гидроксилазы. Изменения наружных половых органов идентичны простой вирильной форме. Если диагноз при рождении не был поставлен, то самое раннее на 2-3 сутки, иногда в течение первых 2-х недель после рождения появляется рвота фонтаном, жидкий стул, признаки дегидратации с последующим развитием коллапса или шока – развивается острая надпочечниковая недостаточность. Необходима неотложная посиндромная терапия. Гипертоническая или гипертензивная форма. В основе её развития лежит недостаточность 11α-гидроксилазы. Клинически эта форма полностью идентична простой или вирильной, но сопровождается и артериальной гипертензией. Редкие формы ВДКН. В случае несоответствия строения наружных половых органов генотипу новорождённого (при генотипе 46XY строение гениталий женского типа) помимо других вариантов нарушения половой дифференцировки необходимо исключить и такие формы ВДКН, как врождённая липоидная гиперплазия и недостаточность 17-гидроксилазы. Пренатальная диагностика стала возможной благодаря развитию техники молекулярно-генетических исследований, но ещё не вошла в широкую медицинскую практику и пока носит экспериментальный характер. При этом путём амниоцентеза в клетках амниотической жидкости или в пробах ворсинок хориона определяют возможность мутации Р450с21, в самой амниотической жидкости определяют уровень не только 17-ОН-прогестерона, но и других метаболитов. Родителей пробанда обследуют с нагрузкой препаратами АКТГ для исключения гетерозиготных носителей. Дата добавления: 2015-02-05 | Просмотры: 1034 | Нарушение авторских прав |