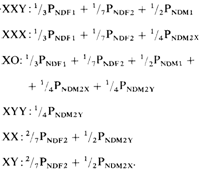|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Х-хромосо иные анеуплоидии у человека: современное состояние проблемыРазличие между Х-хромосомными и аутосомными анеуплоидиями. Вскоре после первых открытий анеуплоидии по половым хромосомам последовали и друше. Если рассматривать все эти случаи в целом как группу, то обнаруживаются заметные отличия от аутосомных анеуплоидии, о которых шла речь ранее: 1) умственное развитие в среднем хотя и ниже нормы, но аномалии развития мозга выражены не столь отчетливо, как при аномалиях аутосом. Многие пробанды имеют нормальное умственное развитие, а некоторые - даже выше среднего (разд. 8.2.2.2); 2) фенотипические нарушения в большей степени затрагивают развитие половых органов и гормонозависимый рост. Наблюдаются и другие пороки развития, особенно при синдроме Тернера, но встречаются они реже и по масштабам менее тяжелые. В конечном счете Х-хромосомная анеуплоидия не нарушает эмбриональное развитие в такой степени, как аутосомные анеуплоидии. Объяснение, по-видимому, состоит в том, что в норме женщина имеет две, а мужчина только одну Х-хромосому, в связи с чем в эволюции сформировался специальный мощный механизм компенсации различий в дозе генов, сцепленных с Х-хромосомой. Этот же механизм оказался фактором, благоприятствующим носителям Х-хромосомных анеуплоидии. Клиническая классификация Х-хромосомных анеуплоидии: мозаики. Наиболее важные численные и структурные аномалии Х-хромосом представлены в табл. 2.10. В общем, число добавочных Х-хромосом увеличивает степень умственной отсталости. Число телец Х-хроматина всегда на одно меньше, 2. Хромосомы человека 101
чем число Х-хромосом. В табл. 2.10 описаны также наиболее частые типы мозаицизма, однако обмены с участием Х-хромосомы не указаны. В последнем случае приложимы те же правила, что и для аутосомных транслокаций, в частности, в отдельных семьях наблюдается значительная вариабельность фенотипических проявлений. Некоторые из Х-аутосомных обменов важны для разработки теоретических концепций относительно инактивации Х-хромосомы. Крупные пороки развития обнаруживаются чаще всего при гонадальном дисгенезе, который является наиболее гетерогенным в клиническом и цитогенетическом отношении. Некоторые авторы, исходя из клинических принципов, предполагают возможность подразделения этого синдрома на варианты [88, 477]. Наиболее известная классификация основана на противопоставлении, с одной стороны, простого гонадального агенеза без добавочных симптомов, а с другой – синдрома Тернера с признаками, представленными на рис. 2.70. Однако онтогенетические данные слабо или вовсе не коррелируют с таким подразделением на две группы. Анеуплоидия ХО встречается много реже, чем варианты XXY и XXX. Ожидаемые частоты различных типов зигот основаны на закономерностях распределения хромосом по гаметам после нерасхождения в первом или втором делениях мейоза у лиц обоего пола (рис. 2.71). При этом делается допущение, что вероятность оплодотворения любой яйцеклетки спермием X или Y составляет 1/2 и что все спермин (включая и анеуплоидные) участвуют в оплодотворении яйцеклеток. Эти события можно обозначить следующим образом: NDF1 нерасхождение в первом мейотическом делении у женщин NDF2 нерасхождение во втором мейотическом делении у женщин NDM1 нерасхождение в первом мейотическом делении у мужчин NDM2X нерасхождение, затрагивающее X-хромосому во втором мейотическом делении у мужчин NDM2Y нерасхождение, затрагивающее Y-хромосому во втором мейотическом делении у мужчин
102 2. Хромосомы человека
Ожидаемые относительные частоты (вероятности, Р) различных типов зигот:
Следовательно, при отсутствии влияния других факторов и при условии равных вероятностей для NDM1 и NDM2X зиготы XXY должны встречаться несколько чаще, чем зиготы XXX. Существуют некоторые доказательства того, что нерасхождение аутосом происходит чаще в первом, чем во втором мейотическом делении (разд. 5.1.2.3). Теоретически зиготы ХО должны встречаться чаще, чем зиготы других типов. Однако эмпирические данные не соответствуют этому, синдром Тернера встречается много реже, чем зиготы XXX и XXY. Это указывает на наличие сильного отбора против гамет, не содержащих половой хромосомы X, и/или на наличие сильного внутриутробного отбора против зигот ХО. Последнее предположение подтверждается тем, что среди спонтанно абортированных зародышей тип ХО встречается относительно часто. Есть и другие доказательства: риск нерасхождения обычно увеличивается с возрастом матери (разд. 5.1.2.2). Для анеуплоидий типа XXX и XXY в отличие от типа ХО эта закономерность выявляется вполне четко. В настоящее время допускается, что выжившие зиготы ХО являются результатом не мейотического, а ми готического нерасхождения или утраты хромосом на ранних стадиях дробления. Об этом же свидетельствуют данные, что в группе больных с синдромом Тернера мозаики встречаются относительно чаще, чем в группах XXX- и XXY-анеусомиков. С другой стороны, зиготы XYY могут возникать только вследствие нерасхождения во втором мейотическом делении у мужчин. Однако они встречаются почти так же часто, как и зиготы XX Y. Следовательно, вероятность нерасхождения по Y-хромосоме, PNDM2Y, явно много выше, чем отдельные вероятности разных типов нерасхождения по Х-хромосоме. Наблюдались мозаики всех типов. Механизмы возникновения мозаиков обсуждаются в разд. 5.1.6. Интерсексы. На основании клинических данных различают три типа интерсексов: 1) истинный гермафродитизм: присутст- 2. Хромосомы человека 103 вуют половые клетки обоих полов; 2) мужской псевдогермафродитизм: имеются только тестикулы; 3) женский псевдогермафродитизм: имеются только яичники. К сожалению, эта простая классификация не совпадает с цитогенетическими данными, поскольку в каждой группе встречаются хромосомные варианты, включая и такие случаи, как, например, 46,ХХ у мужчин. Многие интерсексы являются мозаиками, содержащими клетки с различным набором половых хромосом в разных комбинациях. Например, фенотип мозаиков 45,XX/46,XY может проявиться в виде овариального дисгенеза, гонадального дисгенеза, с мужским псевдогермафродитизмом или в форме «смешанного гонадального дисгенеза», когда одна гонада представлена фиброзным тяжем, а другая – диспластическим тестикулом. Некоторые истинные гермафродиты имеют кариотип 46,XX/46,XY. Мозаицизм типа XX/XY может возникать как следствие любого из девяти различных механизмов, таких, как оплодотворение ооцита двумя различными спермиями, слияние двух оплодотворенных яйцеклеток, митотическая ошибка во время первого дробления или внутриутробный обмен стволовыми кроветворными клетками между разнополыми дизиготными близнецами (разд. 3.8.3). Первичная функция полоопределяющих факторов состоит в индукции гонад. Гонады в свою очередь детерминируют развитие других половых органов и вторичные половые признаки. Нарушения индукции гонад могут быть вызваны либо аномалиями набора половых хромосом, либо другими факторами, не имеющими прямого отношения к половым хромосомам. В последнем случае интерсексы могут иметь нормальный набор хромосом как XX, так и XY. Сбалансированные структурные перестройки с участием Х-хромосомы часто ведут к бесплодию у обоих полов [427]. Y-хромосома как фактор, определяющий мужской пол. Развитие тестикул детерминируется Y-хромосомой. Бюлер [315], обобщив данные многочисленных литературных сообщений о больных с аномалиями половой дифференцировки и структурными аномалиями Y-хромосомы, попытался локализовать различные функции в эухроматическом районе Y-хромосомы человека (рис. 2.72). На этом рисунке отсутствует, однако, информация, полученная на основе использования специфических ДНК-зондов. Различные типы аномалий Xи Y-хромосом весьма информативны относительно механизмов полового развития человека. Однако полезные сведения по этой проблеме можно получить также и при изучении метаболических аномалий с простым типом наследования, из опытов по получению химер у животных, а также из исследований HY-антигена. Кроме того, для полного понимания процесса половой дифференцировки необходимы знания о генетическом контроле регуляторных функций гормонов. В связи с этим особое значение приобретает концепция рецепторных болезней. Обсуждение этих вопросов будет представлено в разд. 4.7.5, здесь же мы обсудим один специальный вопрос – дозовую компенсацию.
2.2.3.3. Дозовая компенсация Х-хромосомы млекопитающих [357] Природа Х-хроматина. Когда Барр и Бертрам (1949) [298] открыли Х-хроматин, относительно его природы были высказаны разные гипотезы. По аналогии с дрозофилой вначале предполагалось, что Х-хроматин состоит из гетерохроматиновых райо- 104 2. Хромосомы человека нов обеих Х-хромосом. Это как будто подтверждалось наблюдением, что Х-хроматин состоит из двух частей. Однако Оно и соавт. (1959) [463; 464] показали, что Х-хроматин формируется только одной Х-хромосомой. В делящихся диплоидных клетках регенерирующей печени крысы на стадии профазы Х-хроматиновое тельце выглядело как одна большая хромосома, сильно конденсированная во всей своей длине, а не как объединение гетерохроматиновых районов двух хромосом. В клетках мужчин такая конденсированная хромосома отсутствовала. Был сделан вывод, что каждое тельце Х-хроматина представляет собой единственную гетеропикнотическую Х-хромосому. Половой диморфизм наблюдается потому, что единственная Х-хромосома у мужчины, как и одна из двух Х-хромосом у женщины, остается эухроматической. Этот вывод был подтвержден у других млекопитающих, а Тэйлор (1960) [525] показал с помощью радиоавтографии с меченым 3Н-гимидином, что женская гетеропикнотическая Х-хромосома в соматических клетках китайского хомячка реплицируется только в конце S-фазы. Результаты Тэйлора были подтверждены исследованиями на многих других млекопитающих. Гетерохроматинизация Х-хромосомы происходит на ранних стадиях эмбрионального развития. Дробящаяся зигота млекопитающих не имеет Х-хроматина. Время его первого появления у различных видов варьирует от бластоцисты до стадии ранней первичной полоски, т. е. от 50-клеточной стадии (у свиньи) до стадии в несколько сотен клеток (у человека) и от предимплантационного до постимплантационного периода. В трофобласте человека Х-хроматин появляется на 12-й день развития, а в собственно эмбрионе на 16-й день. Х-хроматин формируется одновременно во всех клетках эмбриона. Данные об анеуплоидных индивидах, имеющих более двух Х-хромосом, свидетельствуют о том, что только одна Х-хромосома остается в эухроматическом состоянии, тогда как все остальные гегерохроматинизированы. Инактивация Х-хромосомы как механизм дозовой компенсации: гипотеза Лайон. В 1961 г. Лайон высказала предположение, что в разных клетках одного организма гетеропикнотическая Х-хромосома может быть либо отцовского, либо материнского происхождения и что эта хромосома функционально неактивна. Таким образом, ею была сформулирована одна из наиболее плодотворных гипотез в генетике млекопитающих, связавшая морфологию хромосомы с ее функцией. Доказательства этой гипотезы опираются на два рода данных. Во-первых, нормальный фенотип самок ХО у мыши свидетельствует о том, что для полноценного развития ей необходима только одна активная Х-хромосома. Во-вторых, у самок мышей, гетерозиготных по некоторым сцепленным с полом генам, обнаруживается мозаицизм. Так, самки, гетерозиготные по сцепленным с полом мутациям, затрагивающим окраску шерсти, имеют шкурку с пятнами нормальной и мутантной окраски. Этот факт заставляет думать, что мозаичный фенотип в данном случае обязан своим возникновением инактивации одной или другой Х-хромосомы еще в эмбриональном развитии. Эта гипотеза предсказывает, что все гены, локализованные в Х-хромосоме и находящиеся в гетерозиготном состоянии, будут иметь мозаичное проявление, так же будут проявляться и аутосомные гены, транслоцированные на Х-хромосому. Когда фенотип не связан с локальным действием гена, возможны различные типы фенотипических распределений. Следовательно, фенотип будет промежуточным между нормальным и гемизиготным, что приведет к неполной пенетрантности у гетерозигот. В том же году Лайон попыталась на основе своей гипотезы объяснить данные, полученные при изучении заболеваний человека, наследуемых сцепленно с полом: при Х-сцепленном глазном альбинизме у гемизиготного мужчины нет пигмента в эпителиальных клетках сетчатки и глазное дно имеет бледную окраску. У гетерозиготных женщин наблюдается неправильная пигментация сетчатки, с пигментированными и не содержащими пигмента пятнами, так что глазное дно имеет не равномерную окраску, а точечную. На рис. 2.73 изображено такое глазное дно. Лайон предсказала также, что должен существовать мозаицизм и по другим Х-сцепленным генам, в частности по вариантам фермента глюкозо-6-фосфат - дегидрогеназы (G6PD). 2. Хромосомы человека 105 Доказательство инактивации Х-хромосомы по данным генетики G6PD у человека. Окраска шерсти у мыши, контролируемая Х-сцепленными генами, или точечная окраска глазного дна при Х-сцепленном альбинизме у человека представляют собой фенотипические признаки, отдаленные от первичного действия гена многими этапами дифференцировки, следовательно, трактовка происхождения таких фенотипов неоднозначна. Эти наблюдения могут служить основой для гипотезы об Х-инактивации, но их недостаточно для ее доказательства. Критическая проверка такой гипотезы подразумевает использование более простых и менее многозначных ситуаций. Хорошей экспериментальной моделью являются Х-сцепленные гены, первичные эффекты которых можно обнаружить уже на уровне белка. Первым Х-сцепленным локусом, для которого такой анализ стал возможным, оказался ген G6PD. Действительно, Бейтлер (1962, 1964) [302, 303] независимо от Лайон разработал концепцию инактивации Х-хромосомы, исходя из анализа вариантов G6PD у человека. Несмотря на то что женщины имеют две, а мужчины - только одну копию гена G6PD, средний уровень активности этого фермента одинаков у обоих полов, так же как и у индивидов с дополнительными Х-хромосомами (XXX, XXY). Следовательно, должен действовать механизм дозовой компенсации. Если женщина гетерозиготна по электрофоретическим вариантам G6PD, то гипотеза случайной инактивации предсказывает, что в одних клетках будет активна Х-хромосома с нормальным аллелем, а в других-с мутантным. Отсюда следует, что данная отдельная клетка способна экспрессировать только один из двух вариантов фермента. Такой мозаицизм действительно был обнаружен Бейтлером в эритроцитах при помощи остроумного, но непрямого метода [304]. Позже этот феномен был подтвержден многими авторами и различными методами [358, 424]. Один из подходов использует клонирование фибробластов в культуре. В африканской популяции ген G6PD полиморфен и представлен двумя частыми аллелями GdA и GdB. В отдельных клонированных фибробластах негритянки, гете-
розиготной по этим аллелям, обнаружены либо GdA, либо GdB варианты (рис. 2.74), но не оба вместе. При обследовании другой женщины, гетерозиготной по недостаточности G6PD, обнаружили аналогичное явление: в одних клонах активность фермента была нормальной, а в других – очень низкой. Еще одно доказательство теории Х-инактивации получено при изучении лейомиомы матки у женщины, гетерозиготной по вариантам А и В G6PD. В опухолевой ткани неизменно обнаруживали только один из двух мутантных типов, в то время как в нормальной ткани матки обнаружены оба типа. Эти явления возможны только при наличии трех условий. 1. Активен только один аллель. 2. Опухоль начиналась с единственной клетки, т. е. она представляет собой клон единичной клетки. 3. Индивидуальные Х-хромосомы остаются либо активными, либо неактивными в течение всего периода роста опухоли. Эти данные подтверждают не только гипотезу Лайон, но и концепцию моноклонального происхождения опухолей. Эксперименты с отдельными клетками были прове- 106 2. Хромосомы человека
дены и в отношении Х-сцепленных аномалий ферментов, например в отношении недостаточности по гипоксантин-гуанин-фосфорибозилтрансферазе (HPRT). И в этом случае феномен случайной инактивации Х-хромосомы был подтвержден. Генетический дефект по HPRT использовался в качестве модели для изучения механизмов действия гена у человека во многих аспектах, что подробнее будет обсуждаться в разд. 4.2.6. Особенно информативным оказалось обследование женщин, гетерозиготных одновременно по G6PD и по другому Х-сцепленному ферменту, фосфоглицераткиназе (PGK) [303]. Было изучено 56 отдельных клеточных клонов, в 33 из них обнаружены фенотипы GdA и PGK-1, в то время как в 23 остальных - GdB и PGK-2. Если бы инактивация этих двух локусов происходила независимо друг от друга, то следовало бы ожидать появления клонов с фенотипом GdA, PGK-2 или GdB, PGK-1. Другие примеры инактивации Х-хромосомы у человека. Инактивация Х-хромосомы у человека была продемонстрирована для многих Х-сцепленных признаков и с помощью разных методов. Особенно интересна демонстрация мозаицизма сетчатки при цветовой слепоте красно-зеленого типа [309]. Сетчатку женщины, гетерозиготной по цветовой слепоте, освещали узким пучком красного или зеленого света. При этом, как и ожидалось, были обнаружены пятна дефектного цветовосприятия, поскольку сетчатка имела мозаичное строение из нормальных и дефектных клонов. Ангидротическая эктодермальная дисплазия является редким Х-сцепленным заболеванием. У пораженных мужчин нет зубов, потовых желез, имеется гипотрихоз, тогда как у гетерозиготных женщин выявляются участки кожи как нормальные, так и лишенные потовых желез [470]. При хроническом грануломатозе с нарушением функции лейкоцитов (30640) [714] бактерицидная активность гранулоцитов резко снижена: они нормально поглощают стафилококки, но не переваривают их. У гетерозиготных женщин имеются две популяции лейкоцитов – нормальная и аномальная [480, 542]. При других Х-сцепленных заболеваниях также обнаружены явления, предсказываемые гипотезой Лайон [133]. Клетки, в которых вторая Х-хромосома не инактивируется [424, 426]. Тельце Х-хроматина становится видимым примерно на 16-й день, на стадии бластоцисты, и вряд ли функциональная инактивация Х-хромосомы происходит раньше. Если одна из Х- 2. Хромосомы человека 107
хромосом с самого начала была неактивной, то различия в фенотипах нормального мужчины (XY) и больного с синдромом Клайнфельтера (XXY), а также нормальной женщины (XX) и больной с синдромом Тернера (ХО) нуждались бы в ином объяснении, чем предполагаемая Лайон возможность действия генов Х-хромосом до инактивации. Имеются доказательства того, что в ооцитах, так же как и в сперматозоидах, Х-хромосома не инактивируется. У мыши фермент LDH контролируется аутосомным геном, a G6PD, так же, как и у человека, Х-сцепленным геном. В оплодотворенных ооцитах ХО была обнаружена сниженная наполовину (по сравнению с ХХ-ооцитами) активность G6PD, а активность LDH была одинаковой [345]. Отсутствие эффекта дозовой компенсации можно объяснить только исходя из предположения, что в ХХ-ооцитах обе Х-хромосомы являются активными. Одна из систем групп крови человека, система Xg, наследуется сцепленно с полом. Согласно предсказаниям гипотезы Лайон, следовало бы ожидать, что у гетерозиготных женщин имеются две популяции эритроцитов, в каждой из которых клетки несут антиген, детерминированный той Xхромосомой, которая оказалась активной в клетке-предшественнице данной популяции. Однако вскоре стало ясно, что это предсказание неверно, поскольку не было обнаружено двух разных популяций эритроцитов. Дополнительно допускалась еще возможность того, что антиген Xg образуется не самими эритроцитами, а привносится их окружением, например сывороткой. И это предположение было опровергнуто наблюдениями на химерных близнецах (разд. 3.8.3). Речь идет о женщинах, которые в дополнение к своим собственным эритроцитам получили стволовые клетки крови от их дизиготных близнецов во время эмбрионального развития. Одни эритроциты имели антигены О и Xga +, другие были АВ и Xga —. Если бы Xg транспортировался из сыворотки, все клетки имели бы тип Xg – генетически «собственный» тип пробанда. Эта загадка была раскрыта, когда установили, что локус Xg расположен вблизи конца короткого плеча Х-хромосомы и что по крайней мере еще один локус, тесно сцепленный с Xg фермента стероидсульфатазы, также не вовлекается в инактивацию. Иначе говоря, дистальная часть короткого плеча Х-хромосомы человека не инактивируется [486, 487]. Активный район охватывает, по-видимому, сегменты Хр 22.13 и Хр 22.3 [502]. В отличие от всех остальных районов инактивированной Х-хромосомы он реплицируется в ранней S-фазе. Что происходит раньше, инактивация Xхромосомы или образование Х-хроматина? На первый взгляд представляется очевидным, что сначала Х-хромосома конденсируется с образованием Х-хроматина, а затем прекращает функционировать. На самом деле ход событий скорее всего противоположный: сначала происходит инактивация, затем формируется Х-хроматин. Такой вывод следует из того факта, что Xхроматин никогда не обнаруживается одновременно во всех клетках. Например, в клонированных культурах фибробластов примерно 30% клеток не имеют Х-хроматина. Количество клеток с Х-хроматином зависит, по-видимому, от клеточного цикла. Измерения активности G6PD в таких культурах показали, что функциональная инактивация была практически полной и отсутствовало какое-либо соответствие между активностью фермента и числом телец Х-хроматина в фибробластах от индивидов с различным числом Х-хромосом. Точный механизм инактивации является еще предметом исследований [357]. Существуют ли генетические различия в паттернах Х-инактивации? Каттанах (1975) [321] описал у мыши Х-сцепленный ген, который контролирует инактивацию Х-хромосомы (X chromosome controlling element, Хсе). Существует мутантный аллель этого гена «выраженная мозаичность» (high variegation, Ohv), который заставляет Х-хромосому оставаться в активном состоянии. Исходя из этого наблюдения, можно предполагать, что и у человека инактивация Xхромосомы может находиться под генетическим контролем. Неслучайная инактивация должна иногда приводить к появлению гетерозигот с клиническими признаками 108 2. Хромосомы человека
Х-сцепленных рецессивных болезней. Если инактивация происходит достаточно рано во время эмбрионального развития – в то время, когда количество клеток данной ткани еще довольно невелико, – то и в этом случае должны иногда появляться пораженные гетерозиготы. Они являются крайними вариантами, которые образуют «хвост» биномиального распределения всех паттернов инактивации. Однако гипотеза случайной инактивации не предсказывает накопления таких случаев среди сибсов. Тем не менее накопление наблюдалось в случае мышечной дистрофии Дюшенна [451] и в одной семье со сфинголипидозом (болезнью Фабри) [488]. В этой семье девять гетерозиготных дочерей больного мужчины можно было разделить на два класса: в одной группе у четырех дочерей активность α-галактозидазы А достигала 50%, в то время как в другой группе активность составляла 20% (активность определяли в лейкоцитах). Авторы обсуждают гипотезу, согласно которой имеется ген, детерминирующий предпочтительную инактивацию Х-хромосомы с нормальным аллелем. Случаи гетерозиготного проявления мышечной дистрофии Дюшенна можно, вероятно, объяснить таким же образом. Точное определение генной активности у гетерозигот по Х-сцепленным болезням способствует накоплению и обобщению подобных сведений. Х-инактивация и аномальные Х-хромосомы [529]. Когда были описаны первые аномальные Х-хромосомы у человека (изохромосомы по длинному плечу, кольцевые хромосомы или делеции части длинного плеча), правила инактивации казались простыми: всегда инактивируется аномальная Х-хромосома, в клетке остается одна нормальная активная Х-хромосома. Чтобы объяснить столь специфический характер инактивации, были выдвинуты две гипотезы. В соответствии с селекционной гипотезой нормальная и аномальная Х-хромосомы инактивируются случайно, так же как и в случае, когда обе нормальные. Однако клетки с инактивированной нормальной Xхромосомой оказываются генетически несбалансированными в большей степени и поэтому должны иметь более низкую скорость деления, чем те клетки, в которых инактивирована аномальная Х-хромосома и которые являются по сути нормальными. Вторая гипотеза предполагает, что инактивация – облигатное свойство аномальных Х-хромосом [156]. По мере накопления данных об аутосомных или Х/Х транслокациях становилось очевидным, что ни одна из этих гипотез не может быть полностью верной. Существует три группы таких транслокаций: реципрокные сбалансированные, практически все они Х-аутосомного типа, причем общее число хромосом равно 46; несбалансированные Х-аутосомные и Х/Х транслокации, также при наличии 46 хромосом; несбалансированные Х-аутосомные транслокации с общим числом хромосом 45. Мы обсудим данные по первой группе, поскольку для остальных эти данные в основном подтверждают выводы, сделанные для транслокаций первой группы. В большинстве случаев таких транслокаций нормальная Х-хромосома инактивируется. Фенотип проявляется в виде гонадального дисгенеза, иногда в сочетании со слабовыраженными признаками синдрома Тернера. Были описаны семьи, в которых у одного из носителей перестройки инактивирована нормальная, а у другого аномальная Х-хромосома. Например, в одной семье у матери обнаружена сбалансированная транслокация Х/21 (рис. 2.75). Одна транслокационная хромосома состояла из длинных плеч, другая из коротких плеч Х-хромосомы и хромосомы 21 с точкой разрыва вблизи от центромеры (но неясно, с какой стороны). У матери, судя по поздней репликации, инактивированной оказалась нормальная Х-хромосома. В клетках этой женщины присутствовало одно тельце Х-хроматина. В то же время у ее дочери имелась не маленькая, а большая транслокационная хромосома и две нормальные Х-хромосомы. Одна из них была инактивирована, но в отличие от матери транслокационная хромосома тоже была инактивирована. Следовательно, дозовая компенсация достигалась у матери и дочери сходным образом. Однако у дочери инактивация распространялась за пределы Х-хромосомы на транслоцированное длинное плечо хромосомы 21, что привело к появлению дополнительных клинических признаков, сходных с теми, которые иногда описываются при моносомии 21. Этот случай показывает, что гипотеза, согласно которой инактивация определяет- 2. Хромосомы человека 109
ся структурой аномальной Х-хромосомы, в общем случае неверна. В этой и других семьях, в которых был обнаружен только один инактивационный паттерн, клетки оставались генетически относительно сбалансированными. Примечательно то, что у большинства носителей сбалансированных транслокаций с участием Х-хромосомы имеется аномальный фенотип, тогда как носители сбалансированных транслокаций между аутосомами обычно имеют нормальный фенотип. Возможны два объяснения. Либо непрерывность определенного района длинного плеча Х-хромосомы необходима для завершения дифференцировки по женскому типу, и в этом случае дефектный фенотип должен быть связан с явлением, называемым в экспериментальной генетике эффектом положения, либо инактивация одной и той же Х-хромосомы во всех клетках вызывает эффект, зависящий, вероятно, от функциональной гемизиготности рецессивного гена. В разных случаях, вероятно, действует тот или иной механизм. У некоторых больных с несбалансированными транслокациями Х-хромосом обнаруживается двойное тельце Х-хроматина, чего никогда не бывает у нормальных индивидов. Это еще одно наблюдение, относящееся к инактивации Х-хромосомы. Описано мною изохромосом по длинному плечу i(Xq) и в то же время лишь единичные случаи i(Хр), хотя оба типа должны встречаться с одинаковой частотой, так как возникают вследствие аномального деления центромеры [519]. С другой стороны, известны Xq-делеции. Эти факты легли в основу гипотезы, согласно которой предполагается существование инактивационного центра в проксимальной части длинного плеча Х-хромосомы. Если этот центр присутствует в аномальной хромосоме, то она инактивируется. Если два центра, как при некоторых несбалансированных транслокациях Х-хромосомы, то могут образоваться два тельца Х-хроматина. Если центра нет, как в большинстве i(Хр)-изохромосом, инактивация не может произойти и зигота, будучи функционально трисомной по ко- 110 2. Хромосомы человека
роткому плечу Х-хромосомы, становится крайне несбалансированной и неспособной к развитию. Недавно получены данные о том, что инактивационный центр может быть расположен на границе между проксимальным Q-темным и Q-светлым сегментами (~ q13) [530]. По некоторым наблюдениям инактивационный импульс, создаваемый этим центром, может распространяться по длине Х-хромосомы в направлении короткого, но не длинного плеча. На рис. 2.76 показаны различные типы аномальных Х-хромосом, их инактивационный паттерн и фенотипы. Относительно Х-аутосомных транслокаций имеется намного больше сообщений, касающихся мышей, чем человека. Некоторые данные по этому вопросу также подкрепляют гипотезу о возможном существовании инактивационного центра. Имеются, в частности, указания на то, что можно даже усилить или ослабить с помощью отбора степень инактивации аутосомного сегмента, транслоцированного на Х-хромосому. Относительно молекулярных механизмов инактивации Х-хромосомы есть много гипотез, однако пока какие-либо определенные выводы преждевременны [357]. Происходит ли инактивация Х-хромосомы в сперматогенезе? Лифшиц и Линдслей (1972) [420] выдвинули гипотезу, согласно которой Xхромосома инактивируется не только у индивидов, имеющих их несколько, но также и в первичных сперматоцитах во время нормального сперматогенеза. Вполне возможно, что это необходимо для нормального созревания сперматозоидов. Больные мужского пола с синдромом Дауна бесплодны из-за остановки сперматогенеза. При исследовании стадии пахитены в сперматогенезе у больных с синдромом Дауна обнаружено, что дополнительная хромосома 21 конъюгирует с X-Y комплексом [399]. Для объяснения некоторых фактов, полученных на мышах, также необходимо допустить, что мейотическая 2. Хромосомы человека 111
конъюгация аутосом или их плеч со свободной частью Х-хромосомы должна быть обычным явлением, и это может приводить к угнетению инактивации Х-хромосомы и созревания сперматозоидов. 2.2.4. Хромосомные аберрации и спонтанные аборты [413] Частота пренаталъной утраты зигот у человека. Около 15% всех беременностей у человека прерываются диагностируемыми спонтанными абортами, если аборт определять как прекращение беременности до 22-й недели (вес эмбриона 500 г и меньше). Однако доказано, что у человека, так же как и у других млекопитающих, теряется много больше зигот на самых ранних стадиях развития, и часто они имеют тяжелые пороки развития [316; 318; 413]. Согласно недавним оценкам, почти 50% всех зачатий не реализуется в пределах первых двух недель развития, до того, как беременность диагностируется [498]. У человека эта ранняя утрата зигот обычно не распознается. В прошлом в качестве причин большей части выкидышей указывались внешнесредовые факторы, такие, например, как хронический эндометрит, который может ухудшать нормальное питание зародыша. Однако высокая частота аномалий развития у абортусов связана скорее с дополнительными - эндогенными причинами. Когда выяснилось, что именно хромосомные аберрации у человека являются причиной синдромов с множественными пороками развития и сниженной жизнеспособностью, исследователи занялись анализом абортированных эмбрионов. Частота хромосомных аберраций. Уже в 1961 г. были описаны два абортуса с триплоидией [335; 475], а в 1963 г. в первых двух сводках цитогенетических исследований абортов [316; 324] выявилась неожиданно высокая доля эмбрионов с хромосомными аномалиями. В последующие годы на эту тему было опубликовано много работ. В недавнем обзоре собраны данные о 3714 образцах из правильно подобранных групп, причем оказалось, что 1499 (40,4%) имеют хромосомные аберрации [413]. Между этими группами наблюдались значительные колебания в доле аномальных кариотипов, что связано, вероятно, с факторами отбора материала, такими, как материнский возраст, неудачная постановка культур клеток, срок беременности. Последний параметр является, по-видимому, наиболее важным. На рис. 2.77 данные представлены в соответствии со сроком беременности. Чаще всего выкидыши происходят в интервале между 8-й и 15-й неделями. Этот показатель снижается примерно до 5% в последнюю неделю беременности. Относительно низкая частота в первые недели беременности объясняется длительной задержкой аномального эмбриона в матке и тем, что такие ранние беременности часто не распознаются. Принимая 15% как частоту диагностируемых спонтанных абортов среди всех распознаваемых беременностей, антенатальная утрата зигот из-за хромосомных аберраций может быть оценена в 5-6%. Это почти в 10 раз больше, чем частота хромосомных аберраций среди живорожденных (около 0,50,6%; разд. 5.1.2.1). Кроме того, эти цифры не включают случаи утраты зигот перед имплантацией в матку. В настоящее время в опытах на животных четко доказано, что предимплантационные потери могут быть даже больше (разд. 5.2). Очевидно, частота спонтанных абортов на самом деле выше. Ясно также, что спонтанные аборты являются мощным средством ранней элиминации дефектных зигот. Типы хромосомных аберраций у абортированных плодов. С самого начала исследований спонтанных абортов стало ясно, что распределение типов хромосомных аномалий среди них отличается от того, которое наблюдается у новорожденных. Некоторые аберрации, например ХО, встречаются как у новорожденных, так и среди абортусов. Другие, например триплоидии, почти всегда ведут к выкидышу и совместимы с рождением живого ребенка только в исключительных случаях (разд. 2.2.1). Третьи, такие, как трисомия 16, можно обнаружить исключительно у абортированных плодов. Более детальный анализ стал возможен с введением методов дифференциального окрашивания. Кризи и соавт. (1976) [329] опубликовали наиболее обширные данные. 112 2. Хромосомы человека
В 15 больших больницах юго-восточной Англии в период с сентября 1971 г. по апрель 1974 г. был собран и изучен материал от 2607 спонтанных абортусов. Плод или плодный мешок удалось обнаружить при 1767 одноплодных и при 36 близнецовых беременностях, в остальных 804 случаях не было найдено ни плода, ни оболочек. Культивировали клетки 1655 одноплодных образцов, остальные были непригодны из-за крайней мацерации или неправильной обработки до поступления в лабораторию. В 513 культурах не было роста, а в 201 пролиферация обнаружена, но пригодных для анализа метафаз не было. Таким образом, кариотип смогли проанализировать в 941 образце одноплодных спонтанных абортов. Это показывает, что даже в хорошо спланированном эксперименте много материала пропадает из-за непредвиденных технических причин и что искажения в оценке частоты хромосомных аномалий у абортусов неизбежны. При изучении 941 одноплодного абортуса у 287 (30,5%) выявлены хромосомные аномалии. В табл. 2.11 приведены частоты основных типов трисомий. В половине случаев были обнаружены первичные аутосомные трисомий, около одной четверти абортусов оказались Х-моносомиками и одна восьмая - полиплоидами. Остальные были в основном моносомны или несли транслокации. Из 149 первичных аутосомных трисомий или транслокаций 89 идентифицированы с помощью дифференциального окрашивания. Из них в 35 случаях обнаружена дополнительная хромосома 16. Дополнительные хромосомы 21 и 22 встречались примерно в 10% всех трисомий, в то время как добавочная хромосома 2 или хромосома 18 выявлены примерно в 5% каждая. Не обнаружена трисомия по хромосомам 1, 5, 6, 7, 11, 12, 17 или 19. Из 36 образцов близнецовых абортусов удалось кариотипировать по крайней мере одного близнеца в 26 случаях. Хромосомных аномалий обнаружено не было. Таблица 2.11. Частота разных аутосомных трисомий в материале 183 спонтанных абортусов (%) [329]
2. Хромосомы человека 113
Фенотипы абортусов. Имеются значительные различия в фенотипах между плодами с различными хромосомными наборами. Трисомия хромосом 2 и 3, например, не совместима с формированием эмбриона и приводит к образованию пустого зародышевого мешка. Трисомия 9 определяет, повидимому, прекращение или искажение эмбрионального развития, что подтверждается редкими наблюдениями над выжившими, несмотря на тяжелые пороки развития, новорожденными (разд. 2.2.1). Эмбриональное развитие в целом, хотя и с нарушениями, совместимо, по-видимому, со всеми типами трисомии D. С другой стороны, трисомия 16 приводит к тяжелым и ранним нарушениям развития – в большинстве случаев наблюдаются пустые зародышевые мешки и сильно дезорганизованные плоды. Наоборот, трисомия 8 вызывает намного меньше нарушений, что способствует относительно более частому выживанию в постнатальном периоде. Из двух типов трисомии по группе G с более благопотучным развитием совместима трисомия 21 в отличие от трисомии 22. Тем не менее, исходя из собственных и литературных данных, авторы считают, что 60% всех зигот с трисомией 21 абортируются! Очень широкая вариабельность фенотипического проявления обнаружена среди зигот ХО, которые представляют наиболее частый кариотип среди всех исследованных абортусов. Наблюдается широкий спектр фенотипов – от внешне нормальных эмбрионов до пустых зародышевых мешков. Характерно наличие гигромы, то ecть водяночного утолщения тканей, которое имеет место также и у живых новорожденных с кариотипом ХО (разд. 2.2.3). Триплоидия (12 случаев) была найдена у эмбрионов и плодов с различными пороками развития (разд. 2.2.1). В противоположность этому тетраплоидия поч ги всегда ассоциировалась с неповрежденными пустыми зародышевыми мешками, два из них имели аномальную амниотическую полость. Следовательно, такое хромосомное нарушение несовместимо с развитием зародыша.
Еще в одном недавнем исследовании содержатся сведения о 3714 спонтанных абортусах [498]. Более половины аномальных кариотигюв представлены трисомиями, около 20%- моносомиями. 18%- полиплоидиями, 3%-структурными аномалиями, остальные прочими нарушениями. Были обнаружены, хотя и с различной частотой, все типы трисомии, за исключением трисомии 1. Эти частоты превышали ожидаемые, основанные на теоретических расчетах общей частоты численных аберраций (трисомий и моносомий вместе). Если принять, что моносомии и трисомии всех аутосом возникают равновероятно, а ранняя элиминация абортусов происходит с неодинаковой частотой, то около 10-30% всех зигот человека должны нести хромосомные аномалии. В определенной степени эти соображения подтверждаются результатами исследования хромосом в сперматозоидах человека [431; 432]. Из 1000 хромосомных наборов сперматозоидов (33 нормальных мужчин) 8,5% содержали хромосомные аберрации, среди них 5,2% были анеуплоядными. Нуллисомные и дисомные сперматозоиды, которые могут формировать моносомные или трисомные зиготы, образуются примерно с одинаковой частотой, причем представлены все группы хромосом с небольшим избытком аномалий хромосом труппы G. Для анеуплоидий, возникающих во время оогенеза, такие данные отсутствуют. Однако хорошо известно, что нерасхождение во время оогенеза обнаруживается много чаще (или более часто совместимо с возникновением оплодотворенных зигот), чем нерасхождение во время сперматогенеза (разд. 5.1.2). С другой стороны, довольно мало оснований считать, что наблюдения над искусственно оплодотворенными in vitro ооцитами (2/3 и 5 них имеют хромосомные аномалии) могут отражать нормальную ситуацию [294]. Некоторые выводы. Данные исследований хромосом у абортусов позволяют сделать немало выводов. Вклад разных хромосом в распознаваемую утрату всех зигот неодинаковый. Неравномерность этого вклада становится особенно очевидной, когда сравнивают абсолютные и относительные частоты аугосомных трисомии. Это обстоятельство не обязательно указывает на различия в частоте нерасхождения в мейозе или во время ранних делений дробления. Однако наибольший риск нерасхождения имеется, по-видимому, для пяти акроцентрических пар D- и G-групп. Явные раз- 114 2. Хромосомы человека
линия в частоте трисомий по остальным аутосомам могут быть объяснены различным временем гибели зигот. Например, если трисомия хромосомы 1 ведет к гибели зиготы до или во время образования морулы, все трисомии хромосомы 1 останутся нераспознанными. Фенотипическая вариабельность может быть широкой даже среди зигот с одной и той же хромосомной аномалией. Это особенно выражено при сравнении зигот с разными кариотипами. Некоторые, такие, как трисомия 21, совместимы с жизнью. Другие, например трисомия 16, несовместимы даже с ранними стадиями эмбрионального развития и, следовательно, полностью детальны. Сравнение анеуплоидных абортусов, а также тканевых культур от выживших носителей анеуплоидий на различных уровнях биохимического и морфологического анализа может стать важным инструментом изучения генетической регуляции процессов эмбрионального развития. Этот вопрос будет снова обсуждаться в гл. 4, посвященной действию гена (разд. 4.7.4). Дата добавления: 2015-12-16 | Просмотры: 895 | Нарушение авторских прав |