|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Патогенез. В настоящее время считается доказанным, что причиной развивающегося инфаркта миокарда является внутрикоронарный тромбозВ настоящее время считается доказанным, что причиной развивающегося инфаркта миокарда является внутрикоронарный тромбоз, возникающий, как правило, на месте имеющейся атеросклеротической бляшки с поврежденной поверхностью. Доказательствами этому положению послужили исследования английских морфологов M.Davies и A.Thomas (1986). Авторы обнаружили в 74 из 100 вскрытий умерших от ИБС в первые 6 ч от начала симптомов внутрипро-светный тромбоз. Причем все тромбы были в местах разрывов богатых липидами атеросклеротических бляшек, в большинстве случаев тромбо-тические массы проникали через трещину внутрь бляшки и тем самым увеличивали ее размеры. Еще одной классической морфологической работой, свидетельствующей о наличии внутрикоронарного тромбоза при нестабильной стенокардии (НС), является исследование E.Falk, опубликованное в 1985 г. Среди 25 внезапно умерших больных НС внутрикоронарный тромбоз был обнаружен практически у всех. У подавляющего большинства умерших тромбы располагались в местах разрывов бля- шек, имели слоистую структуру, что указывало на различный возраст тром-ботических масс, постепенно суживавших просвет коронарной артерии. Совершенствование ангиографи-ческого оборудования, многочисленные ангиографические исследования больных с нестабильной стенокардией без лечения и с введением тромболи-тических препаратов и, наконец, создание коронароангиоскопических катетеров позволили визуализировать внутреннюю сторону коронарных артерий и подтвердить патогномо-ничность внутрикоронарного тромбоза при нестабильной стенокардии. Наличие общих морфологических признаков в виде поврежденной атеросклеротической бляшки с разрывами ее поверхности и формированием внутрикоронарного тромбоза при инфаркте миокарда с зубцом Q и без, нестабильной стенокардии и осложнениях коронарной баллонной ангиопластики (КБА) способствовало формированию понятия "острый коронарный синдром (ОКС)", в патогенезе которого ведущую роль играют нарушение целости атеросклеротической бляшки и тромбоз коронарной артерии [Fuster V. et al., 1992]. В развитии атеросклеротического процесса большое значение имеет
43 - 4886 повреждение эндотелия. Среди повреждающих гемодинамических факторов рассматривают травматизацию эндотелия потоком крови в разветвлениях артериального русла, особенно выраженную при артериальной гипертонии. Повреждение эндотели-альных клеток обусловлено гиперхо-лестеринемией, гипергликемией, курением, повышенным содержанием катехоламинов, иммунных комплексов, а также инфекцией. На ранних стадиях развития атеро-склеретического поражения в артериях обнаруживают так называемые липидные полосы. Полагают, что стадия липидных полос соответствует динамическому балансу между поступлением и выведением липидов из бляшки. Вероятно, на этом этапе, воздействуя на факторы риска, можно добиться уменьшения поступления липидов в бляшку, способствовать развитию экстрацеллюлярного матрикса и тем самым рубцеванию бляшки. В тех случаях, когда поступление липидов преобладает над выведением, бляшка увеличивается, покрышка истончается. На этой стадии развития бляшка становится легкоранимой, склонной к разрывам. Атеросклеротическая бляшка является основным элементом атеросклероза. В атеросклеротической бляшке выделяют ядро, которое состоит из липидов, ограниченных фиброзной капсулой. Участок бляшки, выступающий в просвет сосуда, называют покрышкой; граничащий с сосудистой стенкой, противоположный, — основанием бляшки, а сегменты покрышки бляшки, переходящие на неизмененную стенку артерии, — плечевой областью бляшки. Ядро бляшки содержит свободный холестерин и его эфиры. Ближе к периферии ядра располагаются так называемые пенистые клетки, являющиеся макрофагами, заполненными липидами. Макрофаги, доставив ли-пиды в ядро бляшки, разрушаются и их содержимое увеличивает ядро бляшки. Плечевые области покрышки бляшки в наибольшей степени подвергаются нагрузке при спазме и дилата-ции артерий, они наиболее тонкие из всей покрышки и именно в плечевых областях чаще всего происходят разрывы бляшек. Бляшки бывают концентрическими, вызывающими фиксированную степень стеноза коронарной артерии, и эксцентрическими, при которых степень стенозирования может варьировать. При коронарной болезни сердца у большинства пациентов бляшки разной формы. При исследовании коронарного русла [Hangar-tner Y.R. et al.] показали, что у больных стабильной стенокардией соотношение концентрических и эксцентрических бляшек составляет соответственно 76 и 24 %. У больных, умерших от ОКС после операции АКШ, эксцентрические стенозы зафиксированы чаще — в 70 % случаев [Freudenberg Н. et al, 1981]. Общепризнанно, что при ОКС эксцентрические стенозы бывают чаще. Разрыв покрышки бляшек определяют рядом физических факторов и его чаще наблюдают в местах истончения фиброзной покрышки бляшки и инфильтрации пенистыми клетками. Эксцентрично расположенные бляшки чаще разрываются в плечевой области. Патологоанатомичес-кое сравнение покрышек интактных и лопнувших бляшек позволило установить, что склонность к разрыву зависит от хронического "стресса" артериальной стенки или так называемой "усталости" покрышки, локализации, консистенции и размеров ядра, а также геометрии бляшки и характеристик потока крови. Разрыв бляшки не является чисто механическим процессом. У больных ОКС анализ атероэктомического материала показал наличие в бляшке участков, богатых макрофагами. Макрофаги способны разрушать экстра-целлюлярный матрикс за счет фагоцитоза и секреции протеолитических ферментов, таких как активаторы плазминогена, металлопротеиназы (коллагеназы, желатиназы, строме-лизины), действие которых ослабляет фиброзную покрышку бляшки и способствуют ее разрыву. Металлопротеиназы и их тканевые ингибиторы участвуют в процессах ремодели-рования сосудов. На культуре макрофагов, полученных из человеческих моноцитов, E.Falk и соавт. (1995), Shah и соавт. (1995) установили, что разрушение фиброзной покрышки атеросклеротической бляшки связано с повышенной активностью интер-стициальной коллагеназы и желатиназы. Таким образом, имеются основания полагать, что металлопротеиназы, содержащиеся в бляшке и моноцитах, участвуют в дестабилизации покрышки бляшки у больных окc. Вход, выживаемость и репликация моноцитов (макрофагов) в бляшке также зависят от эндотелиальных адгезивных молекул (VCAM-1), гемо-токсического белка моноцитов (МСР-1), колониестимулирующего фактора моноцитов (M-CSF) и лим-фоцитарного интерлейкина-2 [Steinberg et al., 1997]. Макрофаги в бляшке подвергаются апоптозу — запрограммированной смерти. По неясной на сегодняшний день причине макрофаги получают сигнал к гибели, после этого в ядре начинают образовываться протеазы, разрушается ДНК и клетка гибнет. Полагают, что апоптоз несет защитную функцию, препятствуя накоплению липидов в сосудистой стенке. Неясно, является ли апоптоз причиной для активации металлопротеиназ, тем не менее это явление приводит к отшнуровыванию поверхностных микрочастиц клеток и экспонированию на их поверхности фосфатидил-серина, что обеспечивает потенциальную прокоагулянтную активность. Отшнуровывающиеся поверхностные микрочастицы макрофагов служат источником тканевого фактора, активность которого в экстрактах бляшки высокая, тканевый фактор активизирует каскад коагуляции при разрыве бляшки. В лопнувших бляшках обнаруживают и другие элементы воспаления, включая тучные клетки и нейтрофи-лы. Тучные клетки находят в небольшом количестве в плечевых областях интактных бляшек. Известно, что тучные клетки секретируют проте-олитические ферменты: триптазу и химазу, которые в свою очередь активируют проферменты металлопротеиназ. Роль нейтрофилов менее понятна, их редко находят в интактных бляшках. Похоже, что они попадают в бляшку вскоре после разрыва ее покрышки. В результате разрыва ранимой бляшки, сопровождающегося изменением ее геометрии и тромбозом, образуется так называемое осложненное поражение. Быстрое изменение геометрии атеросклеротической бляшки при ОКС на ангиограммах проявляется полной или частичной окклюзией коронарной артерии. Довольно часто причиной быстрых изменений в геометрии атеросклеротической бляшки является пристеночный тромбоз, который в дальнейшем может подвергаться организации и участвовать в прогрессии атеросклероза. При разрыве бляшки в формировании и росте тромба принимают участие множество локальных и системных факторов. К местным относят эрозии или изъязвления в покрышке бляшки, изменения в ее геометрии, определяющие степень стеноза артерии, состав (наиболее тромбогенны бляшки, богатые липидами). Важно учитывать и величину поверхности тромба с экспонированными на нем тромбоген-ными белками, определяющими дальнейший рост тромба, а также спастические реакции пораженного сегмента артерии. К системным тромбогенным факторам риска относят содержание холестерина, липопротеина, фибриногена, нарушение фибринолиза (повышение ингибитора тканевого
43* активатора плазминогена первого типа), активацию тромбоцитов и факторов свертывания крови (VII фактор, усиление тромбинообразования). Обсуждается роль инфекционных агентов (Chlamydia pneumoniae, Cytomegalovirus, Helicobacter pylory). Пристеночные тромбы могут частично лизироваться за счет активации эндогенного фибринолиза или замещаться соединительной тканью в результате пролиферации сосудистой стенки. Экспериментальные данные о тромбогенности содержимого бляшек весьма ограничены. Тем не менее при сравнении тромбогенных свойств атеросклеротической бляшки на разных этапах ее развития Fernandez-Ortiz и соавт. (1994), Toschi и соавт. (1997) установили, что в наибольшей степени тромбогенные свойства выражены у липидного ядра, содержащего эфиры холестерина и тканевый фактор. Тканевый фактор (ТФ) представляет собой трансмембранный гликопро-теин, инициирующий каскад коагуляции, который, как полагают, является основным регулятором свертывания, гемостаза и тромбообразования. Тканевый фактор образует высокоаффинный комплекс с Vll/VIIa фактором, комплекс ТФ-VIIa активирует IX и X факторы свертывания, что в свою очередь приводит к образованию тромбина. Анализ атероэктоми-ческого материала больных нестабильной стенокардией подтвердил наличие связей между ТФ и макрофагами [Moreno et al., 1996]. Об исключительной роли ТФ в формировании тромба на поверхности лопнувшей атеросклеротической бляшки свидетельствуют экспериментальные данные [Brown et al., 1986] о том, что применение рекомбинантного ингибитора ТФ (rTFPi) способно существенно ограничить рост тромба на поверхности лопнувшей атеросклеротической бляшки. Появляется все больше доказательств, что моноциты и лейкоциты обладают тромбогенными свойствами, экспрессируя ТФ. Имеются данные Ridker и соавт. (1998) о повышении содержания С-реактивного белка при ОКС. Увеличение уровня холестерина, катехоламинов, курение и, возможно, некоторые инфекционные факторы могут способствовать активированию свертывания крови. Однако примерно у Уз больных, умерших внезапно от коронарной патологии, не находят разрывов в богатых липидами молодых бляшках, а обнаруживают лишь поверхностные эрозии в плотных фиброзных бляшках, существенно суживающих просвет коронарных артерий [Farb et al., 1996]. В этих случаях роль системных тромбогенных факторов, а также гиперкоагуляции представляется особенно важной. Это предположение подтверждают и данные [Dangac et al., 1999] о том, что нормализация уровня холестерина снижает тромбогенные свойства крови у больных с гиперлипидемией. Роль инфекционных агентов в патогенезе атеротромбоза заключается в активации циркулирующих моноцитов, лейкоцитов, повышении синтеза и активации ТФ, тромбоцитов, а также повышении уровня фибриногена. Maseri и соавт. указали на роль спазма коронарных артерий в патогенезе ОКС. Склонность к спазму может быть результатом дисфункции эндотелия в сегменте, расположенном вблизи атеросклеротической бляшки, или нарушения реакции сосуда в месте самой атеросклеротической бляшки. Спазм артерии с поврежденным эндотелием вызывают тром-боксан и серотонин, содержащиеся в тромбоцитах, а также тромбин. Обсуждая роль спазма в патогенезе ОКС, необходимо упомянуть о двух веществах, прямо противоположных по своему действию на тромбоциты и гладкую мускулатуру: тромбоксан А2 и простациклин (рис. 12.1). Они оба являются конечными продуктами метаболизма арахидоновой кислоты. Тромбоксан А2, образующийся в
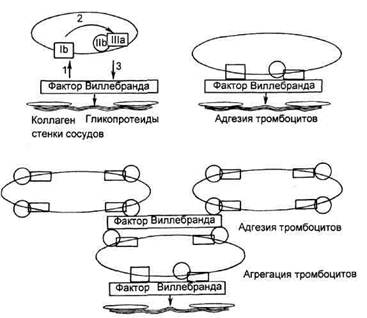
Рис. 12.1. Воздействие тромбоксана А2 и проста-циклина на тромбоциты и гладкую мускулатуру сосудов. тромбоцитах и выделяющийся в кровоток в процессе реакции освобождения, — мощный проагрегант и ва-зоконстриктор. Простациклин образуется в эндотелиальных клетках сосудов и является мощным системным вазодилататором и антиагрегантом. Последнее обусловлено активацией в мембране тромбоцитов аденилат-циклатного механизма, приводящего к увеличению в тромбоцитах содержания циклического АМФ (цАМФ), уменьшению свободного цитоплаз-матического кальция и снижению агрегационной способности тромбоцитов. Импульсом к образованию простациклина эндотелиальными клетками, являющегося веществом, образующимся in situ, могут быть повреждение целости эндотелия, а также появление в кровотоке тромбина. При адгезии тромбоцитов к месту поврежденного сосуда из них выделяется тромбоксан, одновременно с этим из эндотелиальных клеток выделяется простациклин, ограничивая или предотвращая процесс тромбо-образования. С исследований Моп-cada (1976), Vane (1971), посвященных метаболитам арахидоновой кислоты, начался период активного изучения роли тромбоксана и простациклина в патогенезе ИБС и стенокардии. В конце 70-х и 80-х годов была опубликована серия исследований [Sobel, 1981; Tada, 1981; Hirsh, 1981] о роли дисбаланса в соотношении тромбок-сан/простациклин в патогенезе коронарного тромбоза. Группой исследователей во главе с J.Mehta в 1981 г. была выдвинута гипотеза происхождения стенокардии вследствие дисбаланса в имеющемся равновесии тромбоксана и простациклина. При анализе летальных исходов от тромбоза коронарных артерий установлено, что только в 25 % случаев тромбоз связан с эрозией эндотелия, а в 75 % — с разрывом бляшек [Davies, 1990]. Farb и соавт. (1996) полагают, что эрозии эндотелия чаще бывают у женщин. Интересно, что не все разрывы бляшек и связанные с ними тромбозы приводят к клиническим проявлениям острого коронарного синдрома. Исследования показали, что у 17 % больных, умерших от некоронарных причин, находят небольшие свежие разрывы атеросклеротичес-ких бляшек с признаками тромбоза в липидном ядре. Кроме того, Falk (1992) обнаружил у лиц, умерших от разрывов крупных атеросклеротичес-ких бляшек и связанных с ними тромбозов коронарных артерий, в среднем еще 2,5 лопнувшие атеросклеретические бляшки в других местах. Разрывы мелких бляшек способствуют проникновению тромботичес-ких масс внутрь бляшки, стимуляции пролиферации гладкомышечных клеток и дальнейшему росту бляшки. По мнению Davies (1990), Fuster (1994), Flugelman и соавт. (1993), данный механизм лежит в основе развития хронических стенозов и вызывает развитие стабильной стенокардии. Выраженность стеноза в коронарной артерии является важным фактором, определяющим клинические проявления разрывов бляшек. Davies в 1990 г. установил, что у 81 % больных, умерших от тромбоза, развившегося в месте эрозированного эндотелия, были гемодинамически значимые стенозы (> 60 %). Вместе с тем среди лиц, умерших от тромбоза коронарной артерии, сформировавшегося на месте лопнувшей бляшки или изъязвленного поражения, больше половины (60 %) имели гемодинамически незначимые (<60 %) сте- нозы в коронарных артериях. Морфологические данные [Davies, 1990] согласуются с клинико-ангиографи-ческими наблюдениями [Brown et al., 1986; Ambrose et al., 1998] о том, что возникновение нестабильной стенокардии обусловлено ростом бляшек в местах умеренных стенозов. Разрыв бляшек в местах выраженных стенозов не сказывается на коронарном кровотоке, так как длительно существующий стеноз в коронарной артерии способствует развитию коллатерального кровообращения. Вместе с тем разрыв бляшек, умеренно стено-зирующих коронарные артерии, чаще проявляется симптомами ОКС из-за отсутствия развитого коллатерального русла. Разрыв бляшки приводит к экспонированию тромбогенных субстанций субэндотелиальных слоев. В нагруженных липидами макрофагах образуется большое количество тканевого фактора — мощного стимулятора тромбинообразования. Коллаген — мощный стимулятор адгезии и агрегации тромбоцитов — содержится в субэндотелиальных слоях и внутри бляшки. Адгезивный белок — фактор Виллебранда (ФВ) — присутствует и в плазме, и в субэндотелиальных структурах, однако неактивированные тромбоциты могут взаимодействовать только с субэндо-телиальной формой ФВ. Разрыв бляшки приводит к экспонированию субэндотелиального ФВ, что способствует первому этапу образования тромбоцитарного тромба — адгезии тромбоцитов. Адгезия тромбоцитов происходит вследствие связывания ФВ с рецептором мембраны тромбоцитов гликопротеином lb. Фактор VIII — так называемый фактор Виллебранда — представляет собой многомерный белок плазмы, индуцирующий прилипание тромбоцитов к субэндотелиальным структурам. ФВ, помимо ключевой роли в адгезии и агрегации тромбоцитов, участвует в образовании тромба и процессах коагуляции, являясь за- щитным белком для VIII фактора свертывания крови. За последние годы достигнуты большие успехи в изучении ФВ. В результате изучения работ Ruggeri и соавт. (1992), Giusburg и соавт. (1993) изучена структура белка ФВ, идентифицирован его функциональный домен, определены мутации, приводящие к вариантам ФВ. Ген ФВ расположен в 12-й паре хромосом. ФВ имеет в своей структуре цепочку Арг-Гли-Асп — это соединение находят в структуре многих адгезивных белков, которые способны взаимодействовать с так называемыми интегриновыми рецепторами. ФВ синтезируется исключительно в мегакариоцитах, эндотелиальных клетках, находят его и в а-гранулах тромбоцитов, плазме и субэндотели-альном пространстве. В эндотелиальных клетках ФВ находится в тельцах Weibel—Palade и выделяется из них под воздействием различных физиологических стимулов. Основное количество ФВ находится в эндотелии, субэндотелии и тромбоцитах, поэтому его содержание в плазме непостоянно. ФВ выделяется в кровоток в месте и момент повреждения эндотелия и таким образом участвует в регуляции гемостаза. ФВ имеет 2 основные функции: первая — связывание и стабилизация VIII фактора in vivo и in vitro (защита VIII фактора от инактивации протеином С и Ха-фактором); вторая — обеспечение связей между тромбоцитами и сосудистой стенкой (адгезия тромбоцитов) и тромбоцитами (агрегация тромбоцитов) [Меуг, Girma, 1993]. ФВ взаимодействует между компонентами субэндотелия и клеточными рецепторами при высоких скоростях сдвига, т.е. в мелких сосудах и стенозированных артериях, т.е. это единственный белок, осуществляющий адгезию. Последовательность адгезии и агрегации тромбоцитов по Меуг и Girma (1993) представлена на рис. 12.2. ФВ связывается с компонентами субэндотелия, в результате чего в молекуле ФВ происходят кон-
Рис. 12.2. Роль фактора Виллебранда в адгезии и агрегации тромбоцитов. Объяснение в тексте. формационные изменения, способствующие связи участка молекулы ФВ с рецептором мембраны тромбоцита — гликопротеином lb. Таким образом осуществляется начальный (обратимый) процесс адгезии тромбоцитов. Сигнал от взаимодействия ФВ с рецептором lb мембраны тромбоцита передается внутрь клетки и приводит к экспрессии на мембране тромбоцита другого рецептора — гликопротеина IIb/IIIа. Затем осуществляется связь ФВ с рецептором IIb/IIIа и происходят необратимая адгезия и агрегация тромбоцитов. Связь ФВ только с гликопротеином lb недостаточна для полной адгезии тромбоцитов к субэндотелию. Для образования прочной связи, устойчивой к высоким скоростям сдвига, необходима связь ФВ с гликопротеином IIb/IIIа. После прикрепления тромбоцитов к поверхности поврежденного эндотелия они склеиваются друг с другом — так называемый процесс агрегации тромбоцитов. Стимулами к агрегации являются многочисленные аго-нисты, циркулирующие в кровотоке, содержащиеся в атеросклеретической бляшке, субэндотелии, выделяющиеся из тромбоцитов при адгезии и агрегации, а также нарушение текучести крови в стенозированных участках коронарных артерий. Мощнейшими стимуляторами адгезии тромбоцитов являются коллаген субэндотелиального матрикса, а также тромбин. При активации тромбоцитов происходит изменение их формы и активация рецептора ПЬ/Ша. Среди множества стимуляторов активации тромбоцитов (агонистов) следует отметить тромбин, тромбок-сан А2, фактор активации тромбоцитов, серотонин, АДФ, норадреналин. Помимо тромбоцитов, АДФ содержится в эритроцитах, которые и являются основным его источником. АДФ освобождается из эритроцитов при их разрушении в турбулентных потоках, возникающих в суженных атероскле-ротическими бляшками артериях. Каждый агонист, взаимодействуя со специфическим рецептором, образует комплекс и сигнал передается внутрь тромбоцитов при помощи так называемых вторичных мессендже-ров. Считается, что большинство агонистов имеет общий путь передачи сигнала — гидролиз полифосфа-тидилинозитола в диацилглицерол и инозитолтрифосфат, что в последующем вызывает мобилизацию вторичного мессенджера — ионов кальция. Внутритромбоцитарное увеличение кальция способствует сокращению тромбоцитов, секреции АДФ и серо-тонина, активации мембранной фос-фолипазы и арахидонового каскада с образованием ТХА2 — мощного индуктора активации тромбоцитов. Секреция из гранул тромбоцитов биологически активных веществ получила название реакции "освобождения", в результате которой в процесс агрегации вовлекаются новые тромбоциты, он становится самоподдерживающимся и завершается формированием первичного тромбоцитарного тромба. Агрегация тромбоцитов завершается формированием мостиков между адгезивными белками (фибриноген, ФВ) и активированными рецепторами IIb/IIIа тромбоцитов (рис. 12.3). Этот конечный этап агрегации тромбоцитов одинаков при всех возможных стимуляциях тромбоцитов. Структура и функция рецепторов тромбоцитов. Рецепторы тромбоцитов представляют собой гликопроте-ины мембраны, большинство из которых относится к семейству так называемых интегринов [Hynes, 1987]. Интегрины представляют собой ге-теродимерные молекулы, состоящие из семейства а- и b-субъединиц, комбинации которых и являются специфическими рецепторами для различных лиганд. Интегрины находят на поверхностях практически всех клеток, и они участвуют во многих физиологических реакциях. Известно несколько интегринов, участвующих в адгезии тромбоцитов: гли-копротеин Ia/IIA (a2b2) — основной
Рис. 12.3. Агрегация тромбоцитов. рецептор для коллагена; гликопроте-ин Ic/IIа (а5b1) — для фибронектина; а6b1 — для ламинина; Avb3 — для вит-ронектина. Последний рецептор способен узнавать и другие лиганды: фибриноген, фактор Виллебранда, связывающиеся и с рецептором IIb/IIIа. Известно несколько рецепторов, являющихся по структуре не-интегринами и участвующих в адгезии тромбоцитов: гликопротеин IV — рецептор для коллагена и тромбо-спондина, а также гликопротеин lb — связывающий фактор Виллебранда. Таким образом, за процесс адгезии тромбоцитов ответственны несколько рецепторов мембраны тромбоцитов, среди которых есть представители семейства интегринов и неинтегри-нов. Однако основным рецептором, узнающим наибольшее количество лиганд, а именно фибриноген, фибронектин, фактор Вилленбранда и витронектин, и участвующим в процессе агрегации, является гликопротеин IIb/IIIа (aIIb2bз) поверхностной мембраны тромбоцитов. Рецептор тромбоцитов IIb/IIIа — типичный представитель семейства интегринов. Его а-субъединица или гликопротеин IIb (мол. масса 136 kd) состоит из тяжелой и легкой цепей (рис. 12.4). Легкая цепь имеет короткий цитоплазматический хвост, трансмембранную часть и короткий внеклеточный домен. Тяжелая же цепь расположена снаружи клетки. b-Субъединица или гликопротеин IIIа (мол. масса 92 kd) состоит из единственного полипептида (762 аминокислоты) с коротким цитоплазматичес-ким хвостом, трансмембранной частью и большим внеклеточным доменом. Субъединицы нековалентно связаны друг с другом, для сохранения гетеродимерной структуры необходим кальций. Рецепторы IIb/IIIа самые распространенные, на поверхности одного тромбоцита содержится примерно 50 000 рецепторов. Рецепторы тромбоцитов IIb/IIIа изучены в наибольшей степени благодаря исследованиям по изучению тромбастении Гланцмана — врожденной патологии, связанной с отсутствием или резким снижением количества IIb/IIIа-рецепторов. Исследования крови больных тромбас-
Рис. 12.4. Структура Hb/IIIa-рецептора тромбоцитов. тенией Гланцмана, проведенные Plow и соавт. в 1981 и 1985 гг. и Phillips и соавт. в 1988 г. показали, что IIIb/IIIа-рецепторы способны связываться не только с фибриногеном и тем самым осуществлять процесс агрегации тромбоцитов, но и с другими адгезивными гликопротеинами: фи-бронектином, фактором Виллебранда, витронектином, в большей степени отвечающими за адгезию тромбоцитов к субэндотелиальным структурам. Механизм действия IIb/IIIа-рецеп-тора заключается в его способности узнавать 2 характерные аминокислотные последовательности. Первая состоит из аминокислот Арг-Гли-Асп, она обнаружена в фибронектине, фактора Виллебранда, витронектине, а также в а-цепях молекул фибриногена, причем на каждую половину молекулы фибриногена приходится по две ключевых последовательности Арг-Гли-Асп. Следует подчеркнуть, что "ключевая" последовательность Арг-Гли-Асп узнаваема большинством представителей семейства ин-тегринов. Интимные механизмы взаимодействия IIb/IIIа-рецепторов с адгезивными молекулами до конца не изучены, но очевидно, что пептиды или мелкие молекулы, содержащие ключевую последовательность аминокислот Арг-Гли-Асп, могут быть потенциальными ингибиторами взаимодействия IIb/IIIа-рецепто-ров тромбоцитов с фибриногеном. Вторая цепочка аминокислот, узнаваемая IIb/IIIа-рецепторами тромбоцитов, представляет собой Лиз-Глн-Ала-Гли-Асп-Вал, она находится в карбоксильном конце у-цепей фибриногена. В отличие от цепочки Арг-Гли-Асп цепочку Лиз-Глн-Ала-Гли-Асп-Вал обнаружили только в молекуле фибриногена и, вероятно, именно в этом месте фибриноген связывается с IIb/IIIа-рецепторами тромбоцитов. Взаимосвязь между двумя цепочками до конца неясна. Установлено, что пептиды из у-це-пей фибриногена ингибируют связывание фибронектина и фактора Виллебранда с тромбоцитами [Plow et al., 1984]. Это может быть связано с тем, что места на IIb/IIIа-рецепто-рах, узнающие как первую, так и вторую цепочки, частично перекрываются, или же с аллостерическим взаимодействием участков рецепторов, узнающих 2 упомянутые цепочки аминокислот [Plow et al, 1992; Cal-vete, 1994]. Предполагают, что обе цепочки аминокислот взаимодействуют с несколькими участками IIb/IIIа-ре-цептора. При морфологическом анализе коронарных артерий больных, умерших от ОКС, установлено, что в некоторых треснутых бляшках тромбо-цитарные тромбы соединены с ин-тимальным слоем, растут внутрь бляшки, тем самым способствуя увеличению ее размеров. Неокклюзиру-ющие тромбы обычно расположены пристеночно, состоят в основном из тромбоцитов и фибрина и относятся к "белым". На поверхности этих тромбов расположен слой активированных тромбоцитов. В других случаях при нарушении целости бляшки тромб растет внутрь просвета сосуда и может быть неокклюзирующим или окклюзирующим. Финалом активации процесса свертывания на месте лопнувшей атеросклеретической бляшки может быть тромботичес-кая окклюзия сосуда. Тромб, растущий внутрь просвета сосуда и порой окклюзирующий сосуд, в отличие от неокклюзирующего состоит преимущественно из фибрина, эритроцитов, небольшого количества тромбоцитов и условно называется "красным". Итак, патогенез ОКС связан с образованием тромбоцитарного тромба на поверхности лопнувшей или эрозированной атеросклеротичес-кой бляшки. Выраженность ишемии миокарда зависит от степени сужения или окклюзии коронарной артерии, а также ее длительности. Ангио-графические и ангиоскопические исследования свидетельствуют о том, что для НС чаще характерен пристеночный, неокклюзирующий тромбоз, но тем не менее реально уменьшающий кровоток в бассейне пораженной артерии. Возможны преходящие эпизоды тромботической окклюзии длительностью 10—20 мин. Спазм, эндотелиальная дисфункция могут ухудшать коронарный кровоток. При НС находят также эмболии микро-циркуляторного коронарного русла тромбоцитарными агрегатами, приводящие к микроскопическим участкам некроза миокарда. Тромбоци-тарные агрегаты представляют собой скопления активных тромбоцитов с экспонированными IIb/IIIа-рецеп-торами, способных адгезировать к лопнувшим бляшкам в системном кровотоке. При ИМ без зубца Q ангиографи-ческая картина близка к наблюдаемой при НС, внутрикоронарный тромб более устойчивый, периоды окклюзии более длительные (до 1 ч). У 75 % больных ИМ без зубца Q удается выявить ИСА, однако ее окклюзию наблюдают только у 25 % больных. При ИМ без зубца Q кровоснабжение миокарда, расположенного дистальнее окклюзии, осуществляется за счет коллатералей. Принципиальное отличие больных ИМ без зубца Q от больных НС состоит в большей длительности обструкции коронарной артерии, что приводит к некрозу миокарда. В ограничении размеров ИМ играют роль спонтанный тромболизис, устранение спазма, наличие коллатералей. ИМ с зубцом Q отличается развитием быстрой, полной и продолжительной (1 ч и более) окклюзии коронарной артерии. Коронарная артерия окклюзируется хорошо фиксированным, прочным коронарным тромбом. Исходу НС в ИМ, несомненно, способствует сниженный кровоток, создающий повышенную концентрацию тромбогенных факторов in situ. Среди механизмов внезапной коронарной смерти следует учитывать возможность возникновения фатальных ишемических нарушений ритма сердца, связанных с быстрым разрывом бляшки и развитие окклюзирую-щего тромбоза коронарной артерии. С практической точки зрения представляется важным выделение двух клинических вариантов ОКС. Первый характеризуется подъемом сегмента ST на ЭКГ или остро возникшей блокадой левой ножки пучка Гиса, второй депрессией сегмента SТ или динамикой конечной части желудочкового комплекса в виде появления отрицательных зубцов Гили псевдонормализацией зубцов Т. Вы- деление двух вариантов ОКС имеет практическое значение, так как до появления развернутой клинической картины заболевания позволяет определиться с тактикой лечения. Варианту ОКС с подъемом ST соответствуют больные с развивающимся крупноочаговым инфарктом миокарда (ИМ), причиной которого является тромботическая окклюзия коронарной артерии, требующая незамедлительного восстановления кровотока с помощью тромболизиса или коронарной баллонной ангиопластики. Помимо скорейшего восстановления проходимости инф-рактсвязанной артерии (ИСА), анти-тромботическая терапия при ОКС с подъемом ST должна быть направлена на поддержание проходимости ИСА и на борьбу с ее реокклюзией, для чего используются препараты, ингибирующие функцию тромбоцитов, а также образование ключевого фермента свертывания — тромбина и осуществляющие его инактивацию. К ОКС без стойкого подъема ST относятся больные с клиническими признаками нестабильной стенокардии (НС) или мелкоочагового инфаркта миокарда (инфаркта миокарда без Q), характеризующиеся наличием неокклюзирующего тромба на месте лопнувшей атеросклеро-тической бляшки и требующие мероприятий, ограничивающих рост тромба (препараты ингибирующие ключевой фермент свертывания крови — тромбин и функцию тромбоцитов). Дата добавления: 2015-02-06 | Просмотры: 1163 | Нарушение авторских прав |