|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Негели синдром (Негели-Франческетти-Ядасона синдром)Заболевание описано впервые в 1927 г. В 1954 г. Франческетти и Ядасон установили аутосомно-доминантный характер наследования заболевания. Локализация гена известна (17q11.2-q21). Для данного синдрома характерно поражение кожи в виде гипогидроза, сглаженного дерматоглифического рисунка, гиперпигментация периокулярная, периоральная, гиперпигментация груди, шеи, живота. При синдроме Негели патология зубов выражается в изменении их цвета (желтые зубы), сверхкоплектные зубы, аномальная форма зубных коронок, преждевременная потеря зубов, кариес. РАЗДЕЛ 6 АНОМАЛИИ ПРОРЕЗЫВАНИЯ ЗУБОВ Прорезывание зубов у человека происходит в определенной последовательности, причем установлены средние сроки прорезывания каждого зуба с учетом их небольших естественных отклонений в ту или другую сторону. Лишь резкие отклонения от естественных сроков прорезывания зубов рассматримают как аномалию. В табл. 1.15 представлена хронология развития и сроки прорезывания временных и постоянных зубов человека. Первыми в 6-10 мес прорезываются нижние центральные резцы, затем нижние боковые (10-16 мес). Верхние резцы появляются в 8-13 мес; первые моляры - 12-16 мес; клыки - 16-20 мес, вторые моляры - 23-30 мес. Таблица 1-15. Временная динамика развития молочных зубов
Из аномалий развития наиболее часто встречается задержка прорезывания зубов, или ретенция. Помимо генетических факторов, причиной задержки прорезывания зубов могут являтся различные хронические заболевания, эндокринная патология, неполноценное питание (авитаминоз) и др. Реже встречается другая аномалия - раннее прорезывание зубов. При такой аномалии дети уже рождаются с зубами или они прорезываются вскоре после рождения. Такие зубы называются натальными или неонатальными. Натальные и неонатальные зубы затрудняют грудное вскармливание и, как правило, подлежат удалению. Развитие временных и постоянных зубов протекает однотипно, но в разные возрастные периоды. В период, когда временные зубы завершают период своего развития в челюстях, уже имеются закладки постоянных зубов. В ходе прорезывания постоянных зубов происходит выпадение временных. Выпадение временных зубов обычно протекает симметрично на правой и левой сторонах каждой челюсти; у девочек этот процесс происходит раньше, чем у мальчиков. За исключением вторых моляров, зубы нижней челюсти выпадают раньше, чем соответстующие им зубы верхней челюсти. Исследования, проведенные на монозиготных близнецах, показали, что процесс выпадения временных зубов на 80% определяется генетическими факторами. Хронология и периоды формирования постоянных зубов представлена в табл. 1.16. Преждевременное прорезывание зубов является достаточно частым симптомом при многих наследственных моногенных заболеваниях. В настоящее время, по данным каталога МакКюсика, это состояние встречается примерно при 70 формах патологии. Известны заболевания с аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным типом наследования и симптомами раннего прорезывания зубов. Поскольку этот признак не является ведущим в клинической картине многих наследственных заболеваний и не всегда учитывается при клиническом описании, можно предположить, что на самом деле количество наследственных состояний, при которых наблюдается преждевременное прорезывание зубов значительно больше. Практически всегда преждевременное прорезывание зубов сопровождается другими аномалиями зубов и пороками развития, затрагивающими многие органы и системы. Таблица 1-16. Временная динамика развития постоянных зубов
Примечание. * ВУ - внутриутробное развитие. К аномалиям прорезывания зубов относятся: • натальные (прорезавшиеся к моменту рождения) зубы; • неонатальные (у новорожденного, прорезавшиеся преждевременно); • преждевременное прорезывание (раннее прорезывание); • задержка (персистентная) смены (первичных) (временных) зубов; • позднее прорезывание; • преждевременное выпадение первичных (временных) зубов. 6.1. НАТАЛЬНЫЕ, НЕОНАТАЛЬНЫЕ ЗУБЫ Впервые как доминантное состояние описано в 1893 г. (Limrick, 1893). По данным разных авторов, частота натальных/неонатальных зубов у новорожденных 1:1000-3000. По имеющимся данным, некоторые известные исторические личности рождались уже с зубами (король Франции Луи XIV, Ричард III, Ганнибал, Ришелье и др.). 6.1.1. Наследственные заболевания и синдромы с натальными/неонатальными зубами В табл. 1.17 приведен список некоторых известных в настоящее время моногенных заболеваний, сопровождающихся наличием натальных/неонатальных зубов. Зубы, имеющиеся при рождении. Аутосомно-доминантное состояние, когда прослеживается характерное наследование признака без дополнительных аномалий развития. Стеатоцитома множественная с натальными зубами. Кинг и Ли в 1987 г. описали китайскую семью, где в 5 поколениях по меньшей мере у 21 человека отмечались множественные стеатоцитомы в сочетании с неонатальными зубами. Эктодермальная дисплазия с неонатальными зубами. В 1994 г. Turnpenny et al. (1994) описали шотландскую семью с доминантно наследуемой эктодермальной дисплазией и признаками поражения зубов в виде олигодентии, хотя некоторые пораженные имели неонатальные зубы. Пахионихия врожденная, тип 1. Онихогриппоз, гиперкератоз, ороговение участков кожи, гипергидроз ладоней и подошв, стеатоцитома. У больных отмечается умственная отсталость различной сте- Таблица 1-17. Синдромы с натальными/неонатальными зубами
пени выраженности, аномалия волос в виде их истончения, алопеция и др., натальные/неонатальные зубы. Пахионихия врожденная, тип 2. По своим клиническим проявлениям очень похожа на синдром врожденной пахионихии тип 1. Отличием является отсутствие очагов лейкоплакии. Эллиса-ван-Кревельда синдром. Ген заболевания локализован на 4-й хромосоме в районе 4р16. Предположительно заболевание связано с мутацией в гене EVC. Заболевание с аутосомно-рецессивным типом наследования. Характеризуется скелетными дисплазиями, укорочением конечностей, постаксиальной полидактилией, дисплазией ногтей и зубов. Иногда (60%) встречаются сердечно-сосудистые аномалии. Из неврологических проявлений отмечается умственная отсталость и аномалия Денди-Уокера. Среди стоматологических аномалий: неонатальные зубы, задержка прорезывания зубов, частичная олигодентия. Одонто-трихо-ногте-дигитально-ладонный синдром. Mendoza and Valiente (1997) описали предположительно «новый» доминантный синдром эктодермальной дисплазии у двух братьев и их матери. Авторы наблюдали у этих больных натальные зубы, дистрофию волос, патологию пальцев и ладоней, отметили в фенотипе толстые губы, неонатальные зубы, маллоклюзию зубов, аномалии развития кистей и стоп (метакарпальная дисплазия, брахидактилия и др.), дистрофические изменения ногтей и волос. Гиперкератоз контрактуры синдром. Аутосомно-рецессивное заболевание. Локализация гена известна (1p34). Полагают, что заболевание обусловлено мутацией в генах LMNA или ZMPSTE24. Для данного заболевания характерно наличие неонатальных зубов, внутриутробное отставание массы тела, микрогнатия, расщелина губы/ нёба, низко посаженные уши, гипертелоризм, эктропион, атрезия хоан, пороки развития сердечно-сосудистой и мочеполовой системы, скелета, гипоплазия легких, поражение кожи в виде утолщения, гиперкератоза и др. Паллистера-Холла синдром. Аутосомно-доминантное заболевание, вызванное мутацией гена GLI3. Ген расположен в 7p13. Большинство случаев спорадические. Для данного наследственного заболевания характерно наличие неонатальных зубов. Множественные аномалии развития с поражением ушей (микротия, отсутстве наружного слухового прохода и др.), глаз (микрофтальмия). В ротовой полости отмечаются уздечки слизистой щек, мик- роглоссия, расщелина губы/нёба. Пороки сердечно-сосудистой системы, скелета, желудочно-кишечного тракта (неперфорированный анус), мочевой системы (дисплазия и эктопия почек). Пороки развития кистей и стоп (полидактилия, синдактилия, олигодактилия). Врожденные контрактуры. кривошея и злокачественная гипертермия. Редкое наследственное заболевание с аутосомно-рецессивным типом наследования. Проявляется с рождения в виде артрогриппоза, расщелин нёба, наличия зубов при рождении. Для данного заболевания характерным является злокачественная гипертермия в ответ на анастезию. Этот признак меет большое значение в клинической практике. При использовании анестетиков во время стоматологического лечения не исключены осложнения с тяжелыми последствиями. Мекеля синдром, тип 1. Данный наследственный синдром (синоним синдром Грубера, Мекеля-Грубера) - аутосомно-рецессивное заболевание. Локализация гена МКS1в районе 17q23. Это широко варьирующее по клиническим проявлениям заболевание включает в себя почечную патологию с пороками развития центральной нервной системы (энцефалоцеле), патологию печени и полидактилию. Характерны микроцефалия, микрогнатия, микрофтальмия, гипо- и гипертелоризм. К стоматологическим признакам относятся: макростомия, расщелина губы/нёба, натальные зубы. Пороки развития внутренних органов (сердца, печени, селезенки). Пороки развития скелета, половых органов и нервной системы (Арнольда-Киари аномалия, Денди-Уокера аномалия, аплазия мозжечка и др.). РАЗДЕЛ 7 ЗАДЕРЖКА ПРОРЕЗЫВАНИЯ ЗУБОВ Эта аномалия наблюдается в отношении как временных, так и постоянных зубов и встречается в популяции с частотой (1,5%). В большинстве случаев задержка прорезывания связана с неправильным положением зачатков зубов (ретенция). Чаще всего наблюдается в отношении нижних третьих моляров и верхних клыков. Наряду с генетическими факторами причинами ретенции могут являться разнообразные заболевания (рахит, туберкулез, болезни эндокринной системы, аномалии расположения зачатков зубов, аномалии развития челюстей и др.). Эта аномалия является характерной для многих наследственных заболеваний хромосомной и моногенной этиологии. В табл. 1.18 представлены данные о некоторых моногенных заболеваниях и синдромах с задержкой прорезывания зубов. Зубы непрорезавшиеся, с гипоплазией верхней челюсти и вальгусной деформацией коленей. Аутосомно-рецессивное заболевание, характеризующееся аномалиями развития черепа (гипоплазия верхней челюсти и недоразвитие синусов верхней челюсти), деформация коленных суставов, ушных раковин. Зубы: задержка прорезывания временных и постоянных зубов, гипоплазия альвеолярной области. Клейдокраниальная дисплазия. Это заболевание с аутосомнодоминантным типом наследования и с множественными аномалиями скелета, уже рассматривалось ранее в разделе, посвященном наличию сверхкомплектных зубов. Наряду со сверхкомплектными зубами это заболевание характеризуется также задержкой прорезывания временных и постоянных зубов и признаками гипоплазии эмали. Пикнодизостоз. Аутосомно-рецессивное заболевание. Характеризуется низким ростом (взрослые меньше 150 см). Отмечаются микрогнатия, узкое нёбо, задержка прорезывания постоянных и молочных зубов, гиподентия, кариес, скелетные аномалии (аплазия, гипоплазия ключиц, остеосклероз, сколиоз, спондилолистез, брахидактилия, акроостеолиз), дистрофия ногтей (ногти плоские, бороздчатые). Мелника-Нидлса синдром (Мелника-Нидлса остеодисплазия). Х-сцепленное доминантное заболевание, вызвано мутацией гена, кодирующего филамин А (FLNA). Локализация гена Xq28. Заболевание характеризуется задержкой моторного развития, аномалиями лицевой области, микрогнатией, выступающим лбом с низким ростом волос, большими ушами. Часто повторные отиты среднего уха, экзофтальм, гипертелоризм, страбизм. Характерны задержка прорезывания зубов, смещенные зубы. Пороки развития сердечно-сосудистой системы: легочная гипертензия, деформация грудной клетки, укорочение ключиц и лопаток. Скелетные аномалии, аномалии кистей и стоп. Явления акроостеолиза. Патология мочевой системы (гидронефроз, стеноз мочеточников). Нарушение походки. Рутхерфурда синдром. Аутосомно-доминантное заболевание, сопровождающееся патологией глаз (дистрофия радужки) и признаками задержки прорезывания временных и постоянных зубов. Псевдопапилледема, глазной гипотелоризм, блефарофимоз и аномалии кистей. Аутосомно-рецессивное заболевание, характеризующееся множественными аномалиями развития (низкий рост, Таблица 1-18. Наследственные заболевания и синдромы с задержкой прорезывания зубов
Окончание табл. 1-18
микрогнатия, низкопосаженные уши, атрезия наружного слухового прохода, гипотелоризм, эпикант, псевдопаппилодема, блефарофимоз, высокое узкое нёбо, патология скелета - кифосколиоз, деформации пальцев рук и ног). Для данного заболевания также характерны множественные аномалии развития зубов (неправильный прикус, темный оттенок зубной эмали, анодентия, иногда сверхкомплектные зубы, задержка прорезывания зубов). Фиброматоз десен с характерным лицом. Синдром с аутосомнорецессивным типом наследования. Для данного синдрома характерно наличие макроцефалии, гипертелоризм, синофриз, гипоплазия крыльев носа, фиброматоз десен, задержка прорезывания и отсутствие некоторых временных зубов, задержка прорезывания постоянных зубов. Коккейна синдром, тип А. Тяжелое аутосомно-рецессивное заболевание, вызванное гомозиготным состоянием по мутации гена ERCC8. Ген локализован в 5q12. Синдром Коккейна сопровождается нарушениями со стороны центральной и переферической нервной системы (умственная отсталость, деменция, кальцификация базальных ганглиев, дистрофия структур головного мозга, судороги, дисмиелонизация, атаксия, тремор, периферическая нейропатия) и эндокринной систем (гипогонадизм). Патология органа зрения и слуха: пигментная ретинопатия, атрофия зрительных нервов, страбизм, нистагм, катаракта, микрофтальмия, гипоплазия радужки, микрокорнеа, нейросенсорная глухота. Аномалии зубов: кариес, задержка прорезывания, малокклюзия, отсутствие/гипоплазия зубов. Лакримо-ауриколо-денто-дигитальный синдром. Аутосомнодоминантный синдром. Ген локализован в 5p13-p12,4p16.3. Как следует из его названия, основными составляющими этого синдрома являются: патология слезной системы (алакримия, непроходмость слезно-носового канала, аплазия/гипоплазия слезной точки, апла- зия/гипоплазия слезных желез, дакриоциститы), патология слуха (смешанная кондуктивная и нейросенсорная тугоухость), патология зубов (гиподентия, коническая форма зубной коронки, гипоплазия эмали, задержка прорезывания временных зубов). Гиперплазия гипофиза, инсулин-резистентный сахарный диабет и соматические аномалии. Аутосомно-рецессивное заболевание, обусловленное гомозиготным состоянием мутации в гене инсулинового рецептора (INSR), для которого характерна патология эндокринной системы (сахарный диабет, диабетический кетоацидоз),
увеличение гениталий, темная окраска кожи, гипертрихоз. При данной патологии встречаются признаки дисплазии зубов и задержка прорезывания временных зубов. Брахидактилия, тип В1. Аутосомно-доминатное заболевание. Ген идентифицирован (ROR2). Клиническая картина полиморфна. Одно из редких доминантных заболеваний, когда описаны случаи гомозиготного состояния по доминтной мутации. Ведущей симптоматикой являются признаки поражения скелета: сколиоз, патология ребер, конечностей, кистей (гипоплазия/аплазия фаланг пальцев, гипопла- зия/отсутствие метакарпальных костей, симфалангизм, синдактилия, камптодактилия и др.) и стоп (гипоплазия/аплазия дистальных фаланг). Характерным признаком является задержка прорезывания постоянных зубов. Дубовица синдром. Аутосомно-рецессивный синдром, для которого характерными признаками являются: низкий рост (в среднем 44 см) и вес (в среднем 2,3 кг) при рождении, с последующим отставанием в темпах развития. Микроцефалия, маленькое лицо, ассиметрия лица, микрогнатия, оттопыренные, низко расположенные уши. Глазные аномалии: телекант, птоз, блефарофимоз, страбизм, микрофтальмия, тапеторетинальная дегенерация сетчатки, гипоплазия и колобомы радужки. Патология скелета, кожи (экзема и др.). У 10-15% пациентов отмечается умственная отсталость. Иногда лимфомы, нейробластомы, апластическая анемия. Склонность к частым хроническим инфекционным заболеваниям, что связано с
иммунологическими проблемами в виде гипогаммаглобулинемии и снижения уровня IgA. Отмечаются задержка прорезывания зубов, неправильный их рост, кариес. Мукополисахаридоз, тип II. Х-сцепленное рецессивное заболевание, обусловленое мутацией в идуронат-2-сульфатазном гене. Ген локализован в районе Xq28. Для данного заболевания характерными признаками являются: низкий рост (взрослые 120-150 см), макроцефалия, снижение слуха, склонность к хроническим отитам, пигментный ретинит, птоз, макроглоссия, гепатомегалия, пупочные и паховые грыжи, кифоз, гипертрихоз, гидроцефалия, иногда судорожные припадки. Зубы: широко расставленные, поздно прорезывающиеся. Краниометафизарная дисплазия, аутосомно-рецессивная. Макроцефалия, тугоухость, атрофия зрительных нервов, множественные скелетные аномалии (гиперостоз швов черепа, небольшая нижняя челюсть, гипероостоз лицевых костей черепа, облитерация паранозальных синусов и мастоидиты, деформация конечностей), склероз метакарпальных костей и фаланг. Зубы: задержка прорезывания постоянных зубов. Гемифациальная атрофия прогрессирующая. Семейных случаев не описано. Все известные случаи являются изолированными. Характеризуется односторонним поражением лица, с липоатрофией, мышечной атрофией на пораженной стороне лица, одностороннее поражение органа зрения и слуха. Зубы: задержка прорезывания на одной стороне, нарушение прикуса. Чаще в процесс вовлечена левая половина лица. Амело-онихо-гипо-гидротический синдром. Аутосомно-доминантное заболевание. Основными признаками синдрома являются: поражение кожи (себорейный дерматит, гипогидроз, ксероз), поражение ногтей (онихолизис) и зубов (гипокальцифицированная-гипопластичная эмаль), желто-коричневый цвет зубов, задержка прорезывания постоянных зубов. SHORT-синдром. Название синдрома - акроним английских слов, описывающих основные клинические проявления заболевания (Short stature - низкий рост, Hyperextesibility - гиперподвижность суставов/грыжи, Ocular depression - снижение остроты зрения, Rieger anomaly - аномалия Ригера, Teething delay - прорезывание зубов). Аутосомно-доминантноезаболеваниесвышеперечисленнымисимптомами, липоатрофией лица, нормальным интеллектом, инсулин-резистентным диабетом, гипергликемией, нейросенсорной тугоухостью. Патология глаз: миопия, катаракта, глаукома, мегалокорнеа, телекант. Скелетные аномалии: гипермобильность суставов. Отставание костного возраста, большие эпифизы, тонкие диафизы, клинодактилия пальцев рук. Разнообразные варианты патологии зубов: гиподентия, нарушение прикуса, задержка прорезывания зубов. Рахит витамин D-зависимый, тип II. Аутосомно-рецессивное заболевание, обусловленное мутацией витамин D рецепторном гене (VDR). Заболевание с ранним началом (начинается в первые 6 мес. жизни). Некоторые пациенты могут быть вылечены с помощью больших доз витамина D и кальция. Характерными признаками являются: отставание в росте, признаки поражения скелета, ребер «четки». В крови снижение паратиреоидного гормона. Задержка прорезывания зубов, гипоплазия эмали, ранний кариес (с 2 года жизни). Витамин D-зависимый рахит, тип I. Заболевание вызвано мутацией в 25-гидроксивитамин D-3-1-альфа - гидроксилазном гене (CYP27B1). Тип наследования: аутосомно-рецессивный. Характерная клиническая картина формируется на втором году жизни. Помимо типичных фенотипических проявлений, лабораторным подтверждением заболевания является определение в сыворотке крови уровня 1,25-дигидроксивитамина D-3. диагностическими лабораторными показателями являются также гипокальцемия, гипофосфатемия, снижение уровня паратиреоидных гормонов, генерализованная аминоацидурия. Частота этого наследственного заболевания очень высокая (носительство гена 1 на 26) среди канадских французов, проживающих в одном из округов Квебека. Хороший терапевтический эффект оказывает применение 1,25-дигидроксивитамина D-3 или 1-альфагидроксивитамина D-3. DGAPO-синдром. Название синдрома - акроним английских обозначений ведущих клинических признаков (G rowth retardation - отставание в росте, A lopecia -алопеция, P seudoanodontia - псевдоанадентия, O ptic atrophy - атрофия зрительных нервов). При данном заболевании псевдоадентия сочетается с задержкой прорезывания зубов. Эктодермальная дисплазия с кистами надпочечников. Аплазии участков кожи, гипогидроз, ониходисплазия. Эндокринопатия: кисты надпочечников, алактия. Гипоплазия молочных желез и сосков. Зубные аномалии: задержка прорезывания зубов, малые аномалии развития зубов. Паллистера-Киллиана синдром. Этиология заболевания: мозаицизм соматических клеток. При лабораторном исследовании обнаруживается мозаицизм (тетрасомия 12р) в фибробластах кожи. При исследовании лимфоцитов обнаруживаются изохромосомные варианты. Тяжелая патология, вызывающая часто внутриутробную гибель плода. Множественные аномалии развития лица, ушей, глаз, полости рта (макроглоссия, расщелина нёба и др.), сердечно-сосудистой системы, центральной нервной системы, скелета. Стоматологические проблемы выражаются в задержке прорезывания зубов. Миллера-Дикера лиссэнцефалия синдром. Аутосомно-доминанное заболевание, вызванное мутацией в лиссэнцефальном гене 1 (LIS1). При лабораторном исследовании обнаруживается микроделеция хромосомы 17 (район р13.3). Основным способом лабораторной диагностики является флюоресцентная гибридизация in situ критического района хромосомы 17. Для заболевания характерными признаками являются: интра- и постнатальная задержка развития, тяжелая неврологическая сиптоматика (пороки развития мозга: агирия, отсутствие/гипоплазия мозолистого тела, кальцификаты в головном мозгу, ранняя гипотония и позже мышечный гипертонус, отставание в моторном и психическом развитии, умственная отсталость, прогрессирующая спастическая параплегия, судороги и др.). Наряду с неврологической сиптоматикой отмечаются множественные аномалии кистей (полидактилия, клинодактилия, камптодактилия и др.). Характерна задержка прорезывания зубов. Винчестера синдром. Аутосомно-рецессивное заболевание, вызвано мутацией в матриксе металлопротеиназы-2 гене (ММР2). Проявляется в раннем возрасте. Признаки заболевания: низкий рост, гипертрофия десен, контрактуры суставов, артралгии, признаки остеопороза, кифосколиоз, компрессия тел позвонков, патология кистей (остеолизис карпальных костей), стоп (остеолизиз тарсальных костей, анкилоз мелких суставов), гипертрихоз. Задержка прорезывания зубов. Камптодактилии синдром, Гвадалахара тип 1. Аутосомно-рецессивное заболевание, интра- и постнатальное отставание в весе и росте. Микроцефалия, маленькие, низко посаженные, гипопластичные уши. Телекант, микрокорнеа, синофриз, гипертелоризм. Длинная шея. Задержка костного возраста и другие аномалии скелета (деформация грудной клетки, конечностей, брахидактилия, камтодактилия 2-5 пальцев), пигментные невусы, судорожные припадки. К аномалиям развития зубов относятся: олигодентия, шиповидная форма зубной коронки, задержка прорезывания зубов. МОМО-синдром (макростомия, ожирение, микроцефалия, и аномалии органа зрения). Редкий наследственный синдром, вероятнее всего, обусловленный мутацией de novo. Предположительный тип наследования: аутосомно-доминантный. Для данного синдрома характерны большой вес и рост при рождении, ожирение, макрокрания, аномалии органа зрения (колобома сетчатки, нистагм), умственная отсталость. Аномалии зубов в виде нарушения прикуса, тауродонтизма, задержки прорезывания зубов. Сотоса синдром. Точный тип наследования не установлен. Практически все случаи изолированные (известно только несколько семейных случаев). Характеризуется увеличенной длиной и весом тела при рождении (средняя длина 55,2 см, вес 3900 гр). Во взрослом состоянии вес и длина тела чаще нормальные (рост мужчины в среднем 184,3 см, женщины - 172,2 см). Макроцефалия, долихоцефалия. Нарушение слуха (кондуктивная тугоухость), патология глаз (нистагм, страбизм, гиперопия), высокое арковидное нёбо. Встречается патология сердечно-сосудистой системы, скелета (разболтанность суставов, вальгусная деформация, большие кисти и стопы). Патология центральной нервной системы (задержка развития, вариабельная степень умственной отсталости, неонатальная гипотония, гиперрефлексия, судороги, поведенческие проблемы, выраженная задержка развития речи, частичная или полная агенезия мозолистого тела, расширенные желудочки головного мозга). Задержка прорезывания временных зубов.
Наследственная остеодистрофия Альбрехта. Это гетерогенное по своим проявлениям наследственное аутосомно-доминантное заболевание характеризуется низким ростом, ожирением, катарактой, брахидактилией, умственной отсталостью. Отмечаются гипофосфатемия, гипокальцемия, различные эндокринные аномалии. К стоматологическим признакам относятся: врожденная гипоплазия эмали, задержка прорезывания зубов, ана-/олигодентия. Ген заболевания локализован в районе 20q13.2. Акродизостоз. Аутосомно-доминантное заболевание. Низкий рост, укорочение конечностей за счет дистальных отделов. Брахицефалия, гипоплазия верхней челюсти, прогнатизм. Запавшая переносица, короткий маленький нос с открытыми вперед ноздрями, снижение слуха, гипертелоризм, страбизм, голубой цвет радужек, множественные скелетные нарушения: деформации плечевой, лучевой, локтевой костей. Кисти широкие, с укороченными пальцами. Иногда гипогинетализм, гипогонадизм. В 90% умственная отсталость (коэффициент интеллекта 24-85). Задержка прорезывания зубов. Апера синдром. Аутосомно-доминантное заболевание, вызванное мутацией в гене рецепторе фактора роста фибробластов-2 (FGFR2). Нормальные длина и вес при рождении. Характерные аномалии черепно-лицевой области (краниосиностоз, акробрахицефалия, большой поздно закрывающийся родничок, высокий лоб, прогнатизм, гипертелоризм, опущенные вниз глазные щели, атрезия, стеноз хоан, узкое нёбо, иногда расщелина нёба). Аномалии развития скелета, аномалии почек, патология центральной нервной системы (различная степень умственной отсталости, агенезия мозолистого тела, вентрикуломегалия, аномалии лимбической системы). Зубные аномалии: аномальный прикус, задержка прорезывания зубов.
РАЗДЕЛ 8 НАСЛЕДСТВЕННЫЕ АНОМАЛИИ НАРУШЕНИЯ ПРИКУСА Нарушение прикуса может наблюдаться как изолированная аномалия, являться следствием мультифакториальной патологии или внешнесредовых воздействий, либо входить в состав некоторых наследственных заболеваний и синдромов. Как отдельный фенотипический признак аномалии прикуса встречаются в популяции с высокой частотой (табл. 1-19). Таблица 1-19. Популяционные частоты аномалий прикуса (Дистель В.А., 2001)
Наследственные заболевания и синдромы с нарушением прикуса представлены в табл. 1-20. В настоящее время как составляющая часть наследственного моногенного синдрома нарушения прикуса фигурируют более чем при 10 формах аутосомно-рецессивных заболеваний, при 12 формах заболеваний с аутосомно-доминантным типом наследования и при 5 формах Х-сцепленных заболеваний. 8.1. АУТОСОМНО-ДОМИНАНТНЫЕ СИНДРОМЫ С НАРУШЕНИЕМ ПРИКУСА Нарушение прикуса из-за выступающих вперед верхних передних зубов. Заболевание описано у 19 человек в 8 ядерных семьях у трех поколений. Наблюдается нарушение прикуса, выступающие вперед верхние передние зубы. Тип наследования: аутосомно-доминантный. Максилло-назальная дисплазия тип Биндера (Биндера синдром). Синдром описан как самостоятельная нозологическая единица в 1962 г. Binder (1962) описал заболевание как аплазия верхней челюсти, короткий нос с широкой переносицей, аномалия прикуса. Характерной чертой синдрома является гипоплазия срединной части лица. За 20-летний период в Мельбурне (Австралия) было зарегитрировано более 100 пациентов с этим заболеванием. Иногда отмечается гипоплазия терминальных фаланг пальцев, искривление и расщепление ребер. Тип наследования: предположительно аутосомно-доминантный, но не исключена мультифакториальная природа заболевания. Таблица 1-20. Наследственные заболевания и синдромы с нарушением прикуса
Продолжение табл. 1 -20
Окончание табл. 1 -20
Аурикуло-кондилярный (мыщелковый) синдром. Uuspaa (1978) сообщил о заболевании у матери и двух ее сыновей. Патология выражалась в пороках развития наружного уха и гипоплазии нижней челюсти. В дальнейшем появилась информация о клинической вариабельности данного синдрома (без существенного нарушения органа слуха). У 25% пациентов отмечается макроцефалия. Практически всегда микрогнатия, микростомия (52%), глоссоптоз (46%), аномалия нёба (63%), аномалии зубной коронки, нарушение прикуса. Изза орофациальных аномалий: нарушение процесса дыхания, апноэ. Заболевание наследуется по аутосомно-доминатному типу. Одонто-трихо-ногте-пальце-ладонный синдром (OTUDP-син- дром). Тип наследования: аутосомно-доминантный. Как «новый» вариант эктодермальной дисплазии предствлен Mendoza and Valiente (1997). Заболевание было описано у двух братьев их матери и у 18 родственников в пяти поколениях. Авторы наблюдали натальные (при рождении) зубы, дистрофию волос, нарушение прорезывания постоянных зубов, дистрофия ногтей, деформация пальцев, брахидактилию, укорочение метакарпальных и метафизарный костей. Прогнатизм, нарушение прикуса. SHORT-синдром. Название синдрома - акроним английских слов, описывающих основные клинические проявления заболевания (S hort stature - низкий рост, H yperextesibility - гиперподвижность суставов/грыжи, O cular depression - снижение остроты зрения, R ieger anomaly - аномалия Ригера, T eething delay - прорезывание зубов). Аутосомно-доминантное заболевание с вышеперечисленными симптомами, липоатрофией лица, нормальным интеллектом, инсулин-резистентным диабетом, гипергликемией, нейросенсорной тугоухостью. Патология глаз: миопия, катаракта, глаукома, мегалокорнеа, телекант. Скелетные аномалии: гипермобильность суставов. Отставание костного возраста, большие эпифизы, тонкие диафизы, клинодактилия пальцев рук. Разнообразные варианты патологии зубов: гиподентия, нарушение прикуса, задержка прорезывания зубов. Шпринтзен-Гольдберга краниосиностоз (краниосиностоз с арахнодактилией и грыжами). Заболевание вызвано мутацией в фибриллиновом 1 гене FBN1, локализованном в 15q21.1, наблюдается как «новый» наследственный синдром, описано в 1982 г. Shprintzen and Goldberg. Наряду с проявлениями краниосиностоза (микроцефалия, долихоцефалия) отмечаются экзофтальм, телекант, гипертелоризм, страбизм, миопия, птоз, гипоплазия верхней и нижней челюсти, гипертрофия мягких тканей, низко посаженные уши, множественные грыжи, арахнодактилия, камптодактилия. В детстве: гипотония, отставание в развитии, умственная отсталость, псевдорасщелина нёба. Нарушение прикуса. Нунан 1 синдром. Заболевание связывают с наличием мутации в 11 гене протеин-тирозин-фосфотаза нерецепторный тип (PTPN11), локализованном в 12.q24.1. Частота заболевания 1:1000-2500 новорожденных. Тип наследования: аутосомно-доминантный. Для данного заболевания характерными признаками являются: гипертелоризм, опущенные вниз наружные углы глаз, низкий рост, короткая шея, широкая плоская шея и грудная клетка, аномалии сердечнососудистой системы, снижение слуха, моторная неловкость, аномалии пальцев, гипогонадизм, крипторхизм и др. К патологии рта и зубов относятся: высокое, арковидное нёбо, микрогнатия, нарушение прикуса. Акроостеолиз с остеопорозом и аномалиями черепа и нижней челюсти. Аутосомно-доминантный тип наследования. Низкий рост, низко посаженные уши, кондуктивная тугоухость, синофриз, эпикант, телекант, широкий нос, короткая шея, пупочные грыжи, гипоспадия, крипторхизм. Остеопения, патологические переломы, маленькая нижняя челюсть. Гирсутизм, акроостеолизис на кистях и стопах, гидроцефалия. Ранняя потеря зубов, нарушение прикуса.
Трихо-рино-фалангеальный синдром. Заболевание вызвано доминантной мутацией в гене TRPS1. Клинические проявления: грушевидный нос, редкие волосы, конические эпифизы. Низкий рост, микрогнатия, большие оттопыренные уши, деформация проксимальных межфаланговых суставов, укорочение метакарпальных и метатарзальных костей, преждевременный синостоз ростковых зон. Зубные аномалии: маленькие кариозные зубы, нарушение прикуса, задержка прорезывания зубов.
Коккейна синдром, тип В. Заболевание вызвано мутацией гена (ERCC6), локализованного в 10q11. Клинически сходное заболевание с синдромом Коккейна, тип А. Однако синдром Коккейна, тип А, обусловлен мутацией другого гена (ERCC6), локализованного в 5q11. К зубным аномалиям синдрома относят: кариес, задержку прорезывания и формирования зубов, отсутствие и гипоплазию зубов, аномалии прикуса. Акродизостоз. Заболевание было впервые описано Maroteaux and Malamut (1968). Авторы отметили у пациентов признаки акродизостоза (специфическое лицо с коротким носом, открытым ртом и прогнатизмом). Данные аномалии сочетались с маленькими кистями и стопами. Аутосомно-доминантное заболевание. Низкий рост, укорочение конечностей за счет дистальных отделов. Брахицефалия, гипоплазия верхней челюсти, прогнатизм. Запавшая переносица, короткий маленький нос с открытыми вперед ноздрями, снижение слуха, гипертелоризм, страбизм, голубой цвет радужек, множественные скелетные нарушения: деформации плечевой, лучевой, локтевой костей. Кисти широкие с укороченными пальцами. Иногда гипогинетализм, гипогонадизм. В 90% умственная отсталость (коэффициент инеллекта 24-85). Аномалии зубо-челюстной системы выражались в виде нарушения прикуса, задержки прорезывания зубов, гиподентии. Апера синдром (акроцефалосиндактилия, тип 1). Аутосомнодоминантное заболевание, вызванное мутацией в гене рецептора фактора роста фибробластов-2 (FGFR2). Ген локализован в районе 10q26. Впервые заболевание описано в 1906 г. Примерная частота 1:160 000. Исследования, выполненные в Италии, Дании Испании ив4 штатах США, показали, что частота синдрома Апера 15,5: 100 тыс. новорожденных, в Венгрии - 9,9:100 тыс. Средняя частота мутирования гена 7,8Х10-6 за поколоние. Характерными признаками являются нормальная длина и вес при рождении. Специфические аномалии черепно-лицевой области (краниосиностоз, акробрахицефалия, большой, поздно закрывающийся родничок, высокий лоб, прогнатизм, гипертелоризм, опущенные вниз глазные щели, атрезия, стеноз хоан, узкое нёбо, иногда расщелина нёба). Среди всех случаев краниосиностоза синдром Апера составляет 4,5%. Аномалии развития скелета, аномалии почек, патология центральной нервной системы (различная степень умственной отсталости, агенезия мозолистого тела, вентрикуломегалия, аномалии лимбической системы). Зубные аномалии: аномальный прикус, задержка прорезывания зубов. Среди всех случаев заболевания средний возраст матерей составлял 28,9 лет, отцов - 34,1. В 20% случаев рождения детей с синдромом Апера возраст отцов был старше 35 лет. Соотношение пораженных мужчин и женщин 1:1. 8.2. АУТОСОМНО-РЕЦЕССИВНЫЕ СИНДРОМЫ С НАРУШЕНИЕМ ПРИКУСА Нарушение прикуса и низкий рост. Say et al. (1973) описали родных брата и сестру, рожденных в кровнородственном браке. У пациентов отмечались треугольная форма лица, нарушение прикуса и короткий рост (девочка 14 лет была ростом 146 см, мальчик 6 лет - 107 см). Тип наследования: аутосомно-рецессивный. Миастенический синдром врожденный, ассоциированный с недостаточностью ацетилхолинового рецептора. Ген локализован в районе 17p12-p11, 17p13-p12, 11p11.2-p11.1. Тип наследования: аутосомно-рецессивный. Полагают, что заболеваение обусловлено мутацией в эпсилон (CHRNE) и бета CHRNB1 субединиц ацетилхолинового рецептора (AchR). Заболевание может также вызываться мутацией в гене RAPSN. Врожденный миастенический синдром, нейромышечное заболевание. Заболевание и типы мутаций были установлены при исследовавнии цыганских семей, проживавших в Индии и Пакистане. Частота носительства мутантного гена в этой этнической группе составляет 3,7%. Заболевание практически не прогрессирует, характерно выраженное поражение периферической нервной системы. Гипотония, дизартрия. Специфическими фенотипическими признаками синдрома являются: удлиненное лицо, прогнатизм, птоз, офтальмопарез, страбизм, высокое арковидное нёбо, нарушение прикуса, нарушение дыхания из-за слабости дыхательной мускулатуры. У некоторых больных признаки множественного артрогрипоза. При мышечной биопсии отмечается атрофия мышечных волокон 2-го типа. В качестве одного из постоянных признаков является аномалия прикуса. Эктопия хрусталика, краниофациальный дизморфизм. Аутосомно-рецессивное заболевание. До настоящего времени оно описано в нескольких ливанских семьях. Характерные признаки патологии выявляются в виде краниофациальных аномалий, антимонголоидного разреза глаз, нарушения прикуса, эктопии хрусталика, частичной атрофии радужек. Ген не идентифицирован, его локализация неизвестна. Гаррода синдром. Harrod et al. (1977) сообщили о двух братьях с необычным синдромальным поражением в виде умственной отсталости, характерными чертами лица, с большими оттопыренными ушами, гипотелоризмом, длинным носом, маленьким ртом, арахнодактилией, гипогенетализмом, наличием микрокист в корковом слое почек и другими аномалиями развития. Информация о клинических проявлениях синдрома была дополнена описанием 46-летнего пациента с аналогичными признаками и патологией соединительной ткани (мегаколон, варикозное расширение вен). Высокое нёбо, нарушение прикуса. Тип наследования: предположительно аутосомнорецессивный. Остеопетроз с почечным канальцевым ацидозом. Локализация гена 8q22. Sly et al. (1972) описали трех сестер 22, 17 и 15 лет, рожденных от нормальных неродственных между собой родителей. Заболевание характеризуется низким ростом, гепатоспленомегалией, остесклерозом, гипертелоризмом, нарушениями прикуса, анемией. Интеллект нормальный, иногда умственная отсталость. При лабораторных исследованиях отмечается почечный канальцевый ацидоз, периодически гипокалемия. Камптодактилии синдром, тип I, Гвадалахара. Тип наследования: аутосомно-рецессивный. Низкий рост, задержка внутриутробного развития, микроцефалия, брахицефалия, плоское лицо, гипоплазия срединной части лица, эпикант, телекант, микрокорнеа, гипертелоризм, синофриз. Короткий нос, ноздри повернуты вперед, маленький рот с опущенными вниз углами рта, высокое арковидное нёбо, длинная шея. Деформация грудной клетки и другие аномалии скелета. Брахидактилия, каптодактилия, синдактилия. Судорожные приступы, умственная отсталость. Задержка прорезывания зубов, аномалии прикуса. Секкеля синдром. Клинически гетерогенное аутосомнорецессивное заболевание. Ген локализован в области 3q22-q24. Характеризуется отставанием в росте, умственной отсталостью, микроцефалией, судорожными приступами, своеобразным строением головы и лица («птицеголовостью»). Микрогнатия, низкопосаженные деформированные уши, высокое нёбо, иногда ресщелина нёба. Характерна частичная анодентия, гипоплазия эмали, малоклюзия II неправильный рост зубов. Гипоспадия, гипертелоризм, колобома верхнего века и смешанный тип тугоухости. Редкое аутосомно-рецессивное заболевание, выявлено у двух потомков, рожденных в двоюродном браке. Характеризуется низким ростом, прогнатизмом, гипоплазией средней части лица, гипертелоризмом; смешанный тип тугоухости, колобома верхнего века. Гипоспадия, нарушение прикуса. Склеростеоз (кортикальный гиперостеоз с синдактилией). Заболевание вызвано мутацией в гене, кодирующем склерозин (SOST). Ген локализован в 17q12-q21. Аутосомно-рецессивный тип наследования. Характерны большой рост, прогнатизм, гипоплазия срединной части лица, вторичная глухота (из-за гиперостоза черепа), гипертелоризм, птоз, страбизм, нистагм, атрофия зрительных нервов. Скелетные аномалии как следствие гиперостоза. Нарушение прикуса. Псевдопапилледема, глазной гипотелоризм, блефарофимоз и аномалии кистей (акро-ото-окулярный синдром). Тип наследования аутосомно-рецессивный. К настоящему времени имеется описание только четырех пациентов с данным заболеванием. Первая клиническая характеристика заболевания получена при исследовании трех пациентов из двух инбридных бразильских семей. Последнее описание подобного заболевания было в 1997 г. Низкий рост, низкий вес при рождении, микрогнатия, низкопосаженные уши и смешанная форма тугоухости. Псевдопапилледема, гипертелоризм, эпикант, блефарофимоз. Высокое узкое нёбо. Скелетные аномалии: кифосколиоз, аномалии кистей, гипоплазия тенара, гипотенара, кератоз ладоней, укороченные пальцы, частичная синдактилия, укорочение метакарпальных костей. Аномалии стоп: сандалевидная щель, укорочение III-IV пальцев. Множественные пигментные невусы. Зубные аномалии: нарушение прикуса, темноокрашенные зубы, анодентия, сверхкомплектные зубы, аномалии прорезывания зубов. Умственная отсталость, Буэнос-Айрес тип (Мутчник синдром). Аутосомно-рецессивное заболевание. Mutchinick (1972) описал двух пациентов с умственной и физической отсталостью, характерным лицом, пороками развития сердца и почек. В последующем в медицинской литературе появились описания нескольких случаев подобных заболеваний в Японии и Германии. Характерна микроцефалия, прогнатизм, низко посаженные уши, страбизм, миопия, гипертелоризм, птоз. Большой рот, тонкая верхняя губа, высокое арковидное нёбо, нарушение прикуса, кариес. Франк-Тер-Хаара синдром (синдром Мелника-Нидлса). Впервые заболевание было описано у 18-летней арабской девушки, рожденой в кровнородственном браке (1973). Клиническая картина складывается из следующих аномалий: генерализованная костная дисплазия, аномалии скелета (аномалии кистей и стоп, грудной клетки и др.). Характерны: большие глаза, экзофтальм, глаукома, гипертелоризм, микрогнатия, нарушение прикуса, склероз сосцевидного отростка, врожденный порок сердца. Фацио-дигито-генитальный синдром, рецессивный. Аутосомнорецессивное заболевание. Пророрционально низкий рост, брахицефалия, гипертелоризм, выступающая переносица, повернутые вперед ноздри, широкий рот, высокое узкое нёбо, нарушение прикуса. Деформация грудной клетки, маленькие широкие кисти, клинодактилия V пальцев. Халлермана-Штрайфа синдром. Частичная анодентия, наличие неонатальных зубов, малокклюзия, иногда наличие сверхкомплектных зубов. При исследовании полости рта высокое узкое арковидное нёбо, микростомия, тонкие губы и оттопыренная нижняя губа. Характерен комплекс пороков развития. Отмечается низкий вес при рождении, отставание в росте и физическом развитии. Брахицефальная форма черепа, низко посаженные уши, микрофтальмия, катаракты, нистагм, колобомы радужки и диска зрительного нерва, множественные скелетные аномалии. Атрофия кожи, наличие телеангиоэктазов, признаков ксероза. Волосы тонкие, светлые. У 15% больных отмечаются признаки умственной отсталости разной степени выраженности. Горлина-Чаудри-Мосса синдром. Аутосомно-рецессивное заболевание. Характеризуется низким ростом, брахицефалией, гипоплазией срединной части лица, кондуктивной тугоухостью, патологией органа зрения (микрофтальмия, гиперопия, гипертелоризм, птоз). Узкое арковидное нёбо. Скелетные аномалии: краниосиностоз, гипоплазия верхней челюсти и костей носа. Гипертрихоз, гипоплазия дистальных фаланг кистей и стоп. Зубные аномалии: аномалии прикуса, гиподентия, микродентия. Коккейна синдром, тип А. Аутосомно-рецессивное заболевание, вызванное гомозиготным состоянием мутантного гена ERCC8. Ген локализован в 5q12. Характерны нарушения со стороны центральной и периферической нервной системы (умственная отсталость, деменция, кальцификация базальных ганглиев, дистрофия структур головного мозга, судороги, дисмиелонизация, атаксия, тремор, периферическая нейропатия); эндокринной системы (гипогонадизм). Патология органа зрения и слуха (пигментная ретинопатия, атрофия зрительных нервов, страбизм, нистагм, катаракта, микрофтальмия, гипоплазия радужки, микрокорнеа, нейросенсорная глухота). Аномалии зубов: кариес, задержка прорезывания, малокклюзия, отсутствие/гипоплазия зубов. 8.3. НАСЛЕДСТВЕННЫЕ СИНДРОМЫ С НАРУШЕНИЕМ ПРИКУСА, Х-СЦЕПЛЕННЫЕ Дата добавления: 2015-02-06 | Просмотры: 2883 | Нарушение авторских прав |
 Рис. 1.25 а, б. Синдром Коккейна
Рис. 1.25 а, б. Синдром Коккейна Рис. 1.26 а, б. Синдром Дубовица
Рис. 1.26 а, б. Синдром Дубовица Рис. 1.27. Синдром Сотоса
Рис. 1.27. Синдром Сотоса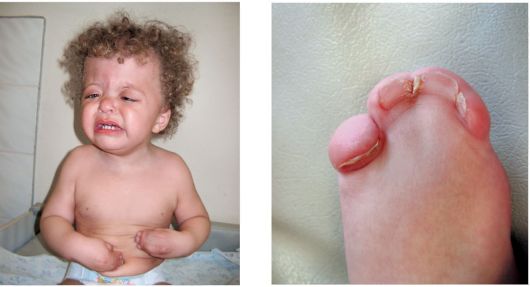 Рис. 1.28 а, б. Синдром Апера
Рис. 1.28 а, б. Синдром Апера Рис. 1.29 а, б. Синдром Нунан
Рис. 1.29 а, б. Синдром Нунан Рис. 1.30. Трихо-рино-фалангеальный синдром
Рис. 1.30. Трихо-рино-фалангеальный синдром