|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Патогенез. С метаболической точки зрения главными признаками ДКА являются накопление органических кислот, ацетоацетата и b-оксибутиратаС метаболической точки зрения главными признаками ДКА являются накопление органических кислот, ацетоацетата и b-оксибутирата, увеличение содержания ацетона в сыворотке и резкое увеличение концентрации глюкозы в крови. С клинической же точки зрения главными угрожающими жизни симптомами являются метаболический ацидоз (вследствие гиперкетонемии), гиперосмолярность — вследствие гипергликемии и потери воды) и дегидратация (вследствие осмотического диуреза, сопровождающего гипергликемию, и рвоты, которая обычно сопровождает тяжелый метаболический ацидоз). Непосредственной причиной всех этих нарушений может служить абсолютная или относительная недостаточность инсулина, развивающаяся в течение нескольких часов или дней. Недостаточность инсулина может быть обусловлена отсутствием его эндогенной секреции (у больных в момент постановки первичного диагноза диабета), неадекватным введением инсулина (у больных с уже диагностированным инсулинозависимым диабетом) или увеличением потребности в инсулине в случаях стресса, связанного с интеркуррентной инфекцией, воспалением или травмой, а также при эндокринных нарушениях. Увеличение потребности в инсулине в таких случаях может определяться повышением секреции гормонов — антагонистов инсулина (например, адреналин, кортизол, глюкагон и гормон роста). Что касается патогенеза гипергликемии, то инсулинодефицитные состояния характеризуются как снижением тканевой утилизации глюкозы, так и избыточной продукцией ее печенью. Если в норме через определенное время после еды скорость высвобождения глюкозы печенью составляет 150—200 мг/мин (2—3 мг/кг в 1 мин), то при ДКА этот показатель достигает 400—600 мг/мин. Таким образом, гипергликемия определяется не отсутствием способности метаболизировать принятые с пищей углеводы, а чрезмерной продукцией глюкозы из эндогенных предшественников. В связи с этим больной диабетом может не есть в течение 12—24 ч, но у него все же развивается гипергликемия; при этом уровень глюкозы в крови превышает 5000 мг/л. Поскольку практически единственными предшественниками синтеза глюкозы de novo являются образующиеся из белка аминокислоты, то такое увеличение глюконеогенеза сопровождается истощением белковых запасов организма и развитием отрицательного азотистого баланса. При ювенильном инсулинозавиоимом диабете повторные приступы диабетического кетоацидоза могут обусловить задержку роста. Однако более непосредственную опасность для больного создает осмотический диурез, сопровождающий тяжелую гипергликемию. Это приводит к дегидратации вследствие потерь воды и натрия с мочой и развитию гиперосмолярности. Одновременно с повышением уровня глюкозы в крови в ней все больше накапливаются кетокислоты (ацетоацетат и b-оксибутират) и ацетон, содержание которых достигает 8—15 моль/л. В течение многих лет развитие гиперкетонемии рассматривали как процесс, который «запускается» и регулируется только скоростью мобилизации жирных кислот из жировой ткани. Однако за последние 10 лет были проведены разнообразные исследования, свидетельствующие о том, что не меньшую роль в регуляции кетогенеза играют изменения печеночного метаболизма, не зависящие от доставки жирных кислот [15]. Как отмечалось, гипоинсулинемия приводит к усилению липолиза в жировой ткани. Это обусловлено выпадением нормального «сдерживающего» влияния инсулина на активность гормон-чувствительной липазы, присутствующей в жировой ткани. Кроме того, снижение поглощения глюкозы жировыми клетками обусловливает нехватку глицерин-3-фосфата, необходимого для реэстерификации жирных кислот in situ. Помимо всего этого, при недостаточности инсулина повышается кетогенная способность печени. Предполагается, что внутрипеченочным метаболическим процессом, определяющим эту активацию кетогенеза, является ацилкарнитинтрансферазная реакция (см. рис. 10—8). Ацилкарнитинтрансфераза катализирует перенос длинноцепочечных жирных кислот через митохондриальную мембрану к месту расположения ферментов, принимающих участие в b-окислении жирных кислот. Ускорение переноса жирных кислот через мембрану приводит, таким образом, к увеличению их окисления и повышению продукции ацетил-СоА. В результате ацетил-СоА становится больше, чем может быть окислено до СО2 в цикле Кребса, что обусловливает конденсацию его молекул с образованием кетокислот. Считают, что механизм, с помощью которого дефицит инсулина повышает активность ацилкарнитинтрансферазы, заключается в. ускорении транспорта карнитина из внепеченочных источников в печень, а также в снижении внутрипеченочного уровня малонил-СоА первого интермедиата в процессе биосинтеза жирных кислот. Малонил-СоА является мощным ингибитором ацилкарнитинтрансферазной реакции. Поскольку дефицит инсулина препятствует синтезу жирных кислот, инсулинодефицитные состояния сопровождаются заметным снижением уровня малонил-СоА. Исследования МсСаrrу и Foster [15] показали также, что не только инсулиновая недостаточность, но и увеличение концентрации глюкагона может значительно увеличивать скорость кетогенеза в печени. Гиперглюкагонемия повышает уровень карнитина, уменьшает концентрацию малонил-СоА и увеличивает активность ацилкарнитинтрансферазы в большей степени, чем это характерно для одной только инсулиновой недостаточности. В результате у панкреатэктомированных больных, у которых отсутствует не только инсулин, но и глюкагон, степень гиперкетонемии оказывается меньшей, чем наблюдаемая при спонтанном диабете [81]. Кроме того, у лишенных инсулина больных ювенильной формой диабета поддержание гипоглюкагонемии с помощью введения соматостатина снижает, хотя я не полностью предотвращает, развитие гиперкетонемии [52].
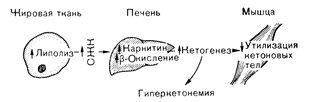
Рис. 10—49. Роль адипоцитов, печеночных и мышечных клеток в развитии гиперкетонемии. Дефицит инсулина приводит к увеличению липолиза в жировых клетках, увеличению накопления карнитина в печеночных клетках (и снижению уровня малонил-СоА), что вызывает ускорение b-окисления жирных кислот (а следовательно, повышение продукции кетоновых тел) и снижению утилизации кетоновых тел в мышечцых клетках. СЖК — свободные жирные кислоты. Помимо увеличения продукции кетоновых тел, гиперкетонемия при диабете определяется снижением утилизации этих органических кислот мышечной тканью. Даже у больных с нерезко выраженной инсулиновой недостаточностью обнаруживается замедление скорости исчезновения кетоновых тел из крови. Сниженная утилизация кетоновых тел служит даже более чувствительным показателем инсулиновой недостаточности, чем избыточная продукция этих соединений [64]. Таким образом, общую последовательность событий, приводящих к развитию гиперкетонемии, можно представить в виде трезубца, изображающего жировую ткань, печень я мышцы (рис. 10—49). Дефицит инсулина приводит к ускорению липолиза в жировой ткани, что обусловливает повышенную доставку свободных жирных кислот в печень. В печени инсулинчувствительное увеличение уровня карнитина и снижение уровня малонил-СоА приводит к стимуляции b-окисления и ускорению продукции кетоновых тел. Последние, высвобождаясь из печени, не могут метаболизироваться с нормальной скоростью в мышечной ткани и поэтому накапливаются в крови. Этот процесс заканчивается метаболическим ацидозом, при котором основными органическими кислотами являются b-оксибутират и ацетоацетат. Кетокислоты обычно присутствуют в плазме в соотношении 3:1 в пользу b-оксибутирата. Вследствие спонтанного декарбоксилирования ацетоацетата образуется и ацетон, концентрация которого в крови может достигать 10—15 ммоль. В силу летучести, выделения легкими и характерного фруктового запаха присутствие ацетона можно определить по дыханию больного, что может служить полезным диагностическим признаком. Помимо изменений в метаболизма глюкозы и жира, для ДКА характерна потеря жидкости и электролитов. Выраженная гипергликемия является причиной осмотического диуреза, приводящего к большой потере с мочой воды и электролитов (по сравнению с плазмой моча при осмотическом диурезе всегда гипотонична в отношении концентрации натрия). Дефицит инсулина может и сам по себе (независимо от осмотического диуреза) обусловливать потерю натрия через почки, поскольку инсулин обладает антинатрийуретическим действием, тогда как острое снижение уровня инсулина приводит к натрийурезу [65]. Если потребление жидкости снижено (что часто бывает вследствие тошноты и рвоты, являющихся ранними предвестниками кетоацидоза), быстро развивается дегидратация; объем крови и периферическое сопротивление сосудов уменьшаются, снижается артериальное давление и нарушается функция почек. Потери воды с рвотными массами и мочой обезвоживают организм. При полном развитии диабетического кетоацидоза дегидратируются все пространства организма и развивается абсолютная недостаточность воды, натрия, калия, магния, хлоридов и гидрокарбоната натрия. Гиповолемия может в свою очередь усиливать кетоз, действуя, вероятно, в качестве стимула секреции адреналина. Наоборот, если больному диабетом, лишенному инсулина, давать жидкость и электролиты, то развитие гиперкетонемии запаздывает или она становится менее выраженной даже если не вводят инсулин [234]. Присутствие большого количества кетоновых тел, являющихся умеренно сильными кислотами, вызывает увеличение концентрации иона водорода в жидких средах организма. В результате происходит уменьшение концентрации гидрокарбоната натрия в сыворотке и увеличивается «анионная брешь» (например, разница между концентрацией натрия в сыворотке и суммой концентраций хлорида и гидрокарбоната натрия превышает нормальную величину 10—15 мэкв/л). Дата добавления: 2015-02-05 | Просмотры: 1146 | Нарушение авторских прав |