|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Генез наиболее важных синдромов при ХПН. 5 страница
Осложнение цитостатической терапии можно разделить на специфические и неспецифические. Некоторые цитостатические препараты вызывают характерные осложнения: винкристин — нейротоксичность (невриты, параличи, атаксия, слепота), синдром неадекватной секреции антидиуретического гормона, алопецию; рубомицин — кардиотоксичность (кардиомиопатия — тахикардия, трофические изменения ЭКГ, одышка, падение артериального давления); аспарагиназа — аллергические реакции вплоть до анафилактического шока, поражения печени (липидоз), поджелудочной железы, в том числе кетоацидоз; циклофосфан — геморрагический цистит, токсический гепатит, синдром неадекватной секреции антидиуретического гормона. Неспецифические осложнения цитостатической терапии могут быть инфекционные и неинфекционные. Неинфекционные осложнения объединяют термином цитостатическая болезнь. Важнейшими признаками ее являются угнетение нормального кроветворения (тяжелые гранулоцитопении, тромбоцитопении и анемии), поражения желудочно-кишечного тракта (стоматит, вызванный как цитостатиком, так и грибами, вирусами, бактериями), цитотоксическая энтеропатия (с преобладанием явлений энтерита или колита), поражения печени (токсико-аллергический гепатит, гепатодистрофия), сердца (кардиомиопатия), легких (цитостатическая пневмопатия, пневмоци-стная пневмония), почек (интерстициальные поражения, обтурация канальцев мочевой кислотой), нервной системы (энцефалопатия, проявляющаяся в виде синдрома апатии, возможно также развитие отека мозга), повышенный риск развития злокачественных опухолей. Для профилактики синдрома лизиса опухоли в момент начала интенсивной цитостатической терапии проводят жидкостную терапию (суточный объем водной нагрузки с учетом выпиваемой жидкости достигает полутора возрастных потребностей) в сочетании с аллопуринолом (суточная доза 10 мг/кг, разделенная на три приема внутрь) и назначением бикарбоната натрия, ибо мочевая кислота лучше растворяется в щелочной среде. Отдаленными последствиями облучения черепа могут быть трудности в учебе, эндокринопатии, включая гипотиреоз, задержку роста, а облучения яичек — нефертильность, которая может потребовать в перспективе даже назначения тестостерона. Симптоматическая терапия. Гемотрансфузии применяют при агрануло-цитозе, сочетающемся с тромбоцитопенией. В этих случаях кровь переливают ежедневно. Оптимально подбирать донора по антигенной системе HLA. Детям с анемией и НЬ менее 70 г/л переливают эритроцитную массу (приблизительно 4 мл на 1 кг массы тела). При глубокой тромбоцитопении (менее 10х109/л) и наличии геморрагического синдрома переливают тромбоцитную массу. Детям с промиелоцитарным лейкозом, учитывая их склонность к ДВС-синдрому, вместе с цитостатической терапией назначают переливания свежезамороженной плазмы, гепарин (200 ЕД/кг в сутки, разделенные на 4 инъекции; по показаниям дозу увеличивают). Детям с глубокой гранулоцитопенией и наличием септических осложнений переливают лейкоцитную массу (вливают 1010 лейкоцитов). Донора подбирают по антигенам HLA. Опасность переливания лейкоцитной массы (как и вообще цельной крови) — развитие реакции «трансплантат против хозяина». В связи с этим мешок с лейкоцитной массой перед введением ребенку рекомендуют облучать дозой 1500 рад. Инфекционные осложнения типичны для больных ОЛ. Оптимально в стационаре следует помещать детей в отдельные боксы или палаты при строжайшем соблюдении правил асептики и антисептики. Любое повышение температуры тела рассматривают как признак инфекции. Антибиотики до выделения возбудителя назначают исходя из установленного фактора широкого распространения у больных условно-патогенной флоры. Профилактическое назначение системных антибиотиков не рекомендуют. Новые методы лечения больных ОЛ. прежде всего, касаются различных аспектов трансплантации костного мозга, что особенно важно для больных ОнЛЛ, у которых часто в процессе лечения возникает аплазия костного мозга. Трансплантируют аллогенный костный мозг с удаленными Т-лимфоцита-ми или очищенный аутологичный костный мозг. Аллогенный костный мозг, совместимый по основным HLA-антигенам, пересаживают сразу по достижении первой ремиссии. Аутогенный костный мозг больного забирают сразу по достижении ремиссии, обрабатывают его моноклональными антителами с иммуноцитотоксинами (например, с рицином) и фармакологическими препаратами (например, гидропероксициклофосфамидом) и вводят больному. Разрабатывают методы сочетания химиотерапии и трансплантаций костного мозга или стволовых кроветворных клеток с предварительным введением колониестимулирующих факторов — гранулоцитарного (Г-КСФ) или грану-ломакрофагального (ГМ-КСФ). ГМ-КСФ, введенный за два дня до начала химиотерапии и далее вводимый в момент ее проведения, способствует увеличению числа и длительности ремиссий при ОЛЛ. Г-КСФ и ГМ-КСФ эффективны И при цитостатической болезни, агранулоцитозе. Самой сложной задачей при трансплантации костного мозга (ТКМ) остается поиск HLA-совместимого донора (наиболее рационален донор-сибс, то есть брат-сестра больного). В конце Прошлого (XX) века было доказана целесообразность трансплантации вместо костного мозга стволовых кроветворных клеток (СКК). В среднем в костном мозге содержится 1 СКК на 105 клеток. Из одной СКК образуется около 1000 клеток-предшественниц и 106 зрелых клеток [Новик А. А. и Богданов А. Н., 2001]. Разработаны методы получения СКК из костного мозга и периферической крови. Наибольшее количество стволовых клеток в костном мозге находится у плода, а в периферической крови человека имеется при рождении. Количество СКК, полученное из плацентарной крови при рождении — достаточно для трансплантации ребенку до 40 кг. Поэтому в настоящее время нередки случаи запланированной беременности для помощи в источнике СКК для старшего ребенка в семье, которому необходима ТКМ. Согласно А. А. Новику и А. Н. Богданову (2001), ТКМ и ТСКК существенно улучшает прогноз при лечении лейкозов, особенно ОМЛ и ХМЛ (табл.164). Согласно данным этих же авторов, в мире ежегодно производят около 50 000 ТКМ и ТСКК. Разрабатываются и иммунологические методы лечения: введение а-интер-ферона (эффективно лишь при волосато-клеточном ОЛ), интерлейкина-2, вакцинация BCG (по схеме!). Диета больным острым лейкозом необходима высококалорийная с полуторным по сравнению с возрастными нормами количеством белков, витаминизированная, богатая минеральными веществами (стол 10а). При назначении глюкокортикоидов рацион обогащают продуктами, содержащими много солей калия и кальция. Деонтологические аспекты очень важны при ведении ребенка, больного ОЛ, и беседах с его родителями. При ребенке никогда не следует называть диагноз. Учитывая психологическую и физическую травматичность современных схем лечения, важно подготовить ребенка и родителей к тем или иным процедурам. Родителям следует сообщить диагноз, как только он станет бесспорным,
J Таблица 164 Пятилетняя безрецидивная выживаемость после ТКМ и ТСКК при ОМЛ (Новик А. А. и Богданов А. Н., 2001).
но одновременно вселять в них оптимизм, разъяснив возможности современной терапии. Необходимо быть очень внимательным к родителям, их вопросам, просьбам. Режим больного определяется его состоянием и гематологическими данными. Диспансерное наблюдение. Осуществляется гематологом специализированного центра и участковым педиатром. Учитывая, что больной практически все время получает цитоста-тическую терапию, необходимо не реже 1 раза в 2 нед делать анализ крови. При поддерживающей терапии цитостатики вводят один раз в неделю, перед этим необходимо подсчитать число лейкоцитов, так как если их менее 1000 в 1 мкл (1х109/л), то препараты не применяют. Дают медикаменты, способствующие увеличению количества лейкоцитов (экстракт элеутерококка по 1 капле на год жизни 2 раза в день, нуклеинат натрия, дибазол, пентоксил, мета-цил), и лишь при увеличении числа лейкоцитов более 1,5х109/л возобновляют цитостатическую терапию. Миелограмму делают перед и после каждого курса реиндукции, которые проводят в стационаре. Нежелательно изменение климатических условий. Ребенка освобождают от профилактических прививок, занятий физкультурой. Его необходимо оберегать от физических нагрузок, психических травм, охлаждения, случайных инфекций. Занятия по школьной программе не противопоказаны, но лучше заниматься дома, так как в школе, особенно в зимне-весенний период, часты ОРЗ среди детей. Прогноз К сожалению, по клинике в момент постановки диагноза ОЛ не всегда можно с уверенностью говорить о прогнозе. Среди больных ОЛЛ выделяют
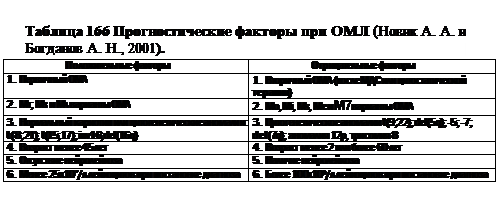
пу детей стандартного риска с благоприятным, как правило, прогнозом и группу больных высокого риска (чаще всего с неблагоприятным прогнозом) — табл. 165. Таким образом, наилучший прогноз при ОЛЛ у больных группы стандартного риска с отсутствием маркеров В- и Т-клеток на лимфобластах, но имеющих ОЛЛа (общий антиген для ОЛЛ). Вообще у 95% всех детей с ОЛЛ достигают ремиссии к концу первого месяца терапии. У 75-95% больных с 0-клеточным вариантом и ОЛЛа-положительных при рациональной терапии ремиссия длится 5 лет, при отсутствии ОЛЛа ремиссия такой длительности бывает лишь у 60%, а при пре-В и В-вариантах, Т-клеточном ОЛЛ переживают 5 лет лишь 40-50% больных. Примерно такой же процент 5-летнего выживания при ОнЛЛ. При этом, чем более зрелые клетки выявляют при ОнЛЛ, тем хуже прогноз. В настоящее время по данным мировой литературы вероятность излечения ОЛЛ составляет, по крайней мере, 50-70%, ОМЛ —15-30%. Вероятность излечения меньшая при обнаружении у больного любых видов транслокаций (табл. 166).
Обычно рецидивы ОЛ наступают в первые 2 года после достижения первой ремиссии. Считают, что если первая ремиссия продолжается у девочек 2 года и у мальчиков 3 года, то можно ставить вопрос о прекращении терапии. При этом проводят цитогенетические и другие методы исследования (поиски онкогенов, клеток с маркерами АЛЛа и др.). Специфические поломки хромосом, онкогены в настоящее время выявляют сравнительно дешевым и нетрудоемким способом, с помощью полимеразной цепной реакции. На фоне поддерживающего лечения или после его окончания возможно появление других опухолевых заболеваний — В-клеточной лимфомы и др. Описаны случаи рецидива ОЛЛ через 4 и даже 9 лет после отмены терапии, то есть говорить о выздоровлении ребенка, перенесшего ОЛЛ, даже после окончания специфической терапии следует очень осторожно.
Хронический миелолейкоз (ХМЛ). Заболеваемость ХМЛ составляет в год 0,12 на 100 000 детей, то есть ХМЛ составляет 3% от всех лейкозов у детей. Ювенильный тип обычно появляется у детей до 2-3 лет и характеризуется сочетанием анемического (бледность, слабость, потеря аппетита и др.), геморрагического, интоксикационного (фебрилитет, боли в костях и др.), про-лиферативного (выраженная' лимфаденопатия, гепатоспленомегалия) синдромов. В анамнезе, а нередко и при поступлении в клинику отмечают ксантомные и экзематозные высыпания (чаще на лице). При анализе крови обнаруживают разной степени выраженности анемию (с тенденцией к макро-цитозу), тромбоцитопению, увеличение СОЭ и лейкоцитоз с резким сдвигом вплоть до миелобластов (от 2 до 50% и более) с наличием всех переходных форм (промиелоциты, миелоциты, юные, палочкоядерные), выраженный мо-ноцитоз. Лейкоцитоз обычно составляет от 25 до 80 х 109/л. В костном мозге — повышенная клеточность, угнетение мегакариоцитарного ростка; процент бластных клеток невелик и соответствует таковому в периферической крови, но все они с признаками анаплазии (диссоциация созревания ядра и цитоплазмы, вакуолизация цитоплазмы, складчатость структуры хроматина и др.). Цитохимические реакции положительные на липиды и пероксидазу в бластных клетках; активность щелочной фосфатазы снижена (так же как и в ней?рофилах, что является одним из аргументов при дифференциации от лей-кемоидных реакций при инфекциях, когда активность щелочной фосфатазы В нейтрофилах повышена). Характерными лабораторными признаками при ювеннльной форме являются также отсутствие Ph '-хромосомы в культуре клеток костного мозга, высокий уровень фетального гемоглобина (30-70%), что отличает эту форму от взрослого типа миелоидного лейкоза у детей. В культуре клеток крови рост моноцитарных колоний. У 55% детей выявляют отсутствие одной из 7-й пары хромосом. Взрослый тип ХМЛ. Иногда диагностируют при плановых осмотрах, при анализах крови у детей школьного возраста, то есть болезнь развивается постепенно. Взрослый тип ХМЛ встречается вдвое чаще, чем ювенильный тип ХМЛ. Считают, что примерно 40% больных ХМЛ в момент постановки диагноза не имеют никаких клинических симптомов, и у них диагноз ставят лишь гематологически. У 20% больных наблюдается гепатоспленомегалия, у 54% — только спленомегалия. Иногда ХМЛ начинается с потери массы тела, слабости, лихорадки, ознобов. Различают три фазы ХМЛ: 1) медленная, хроническая (длится около 3 лет); 2) акселерации (длится около 1-1,5 лет), но при соответствующем лечении заболевание можно вернуть в хроническую фазу; 3) финальная (терминальное обострение, фаза быстрой акселерации, продолжающаяся 3-6 мес и обычно заканчивающаяся смертью). В период акселерации развернутой клинико-гематологической картины заболевания обычно отмечают общее недомогание, повышенную утомляемость, слабость, увеличенный живот, боли в левом подреберье, болезненность при постукивании по костям. Селезенка обычно очень больших размеров (рис. 90). Гепатомегалия менее выражена. Лимфаденопатия обычно минимальна. При анализе крови находят умеренную анемию, нормальное или повышенное количество тромбоцитов и гиперлейкоцитоз (обычно более 100 х 109/л). В лейкоцитарной формуле доминируют промиелоциты, миелоциты, но есть
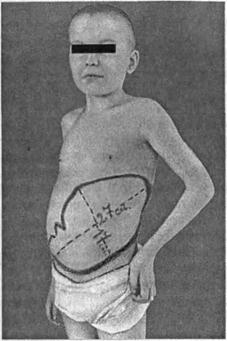
как миелобласты (около 5-10%), так и метамиелоциты, палочкоядерные и сег-ментоядерные формы, то есть отсутствует лейкемическое зияние. Много форм эозинофильного и базофильного ряда, лимфопения, СОЭ увеличена. В костном мозге на фоне повышенной клеточности отмечается незначительное повышение бластных элементов, выраженная мета-миелоцитарная и миелоцитарная реакции. При кариотипировании у 95% больных обнаруживают дополнительную маленькую хромосому в группе 22-й пары — так называемая филадельфийская хромосома (Ph'-хромосома) — результат сбалансированной транслокации материала между 9-й и 22-й хромосомами t (9; 22) (q34; qll). При этой транслокации происходит перенос протоон-когена, называемого с-аЫ, из обычного положения на хромосоме 9 в новое мес-Рис. 90. Спленомегалия у больной то на 22-й хромосоме, называемое Ьсг. В хроническим миелолейкозом. результате образуется новый химерный ген bcr/abl, что приводит к повышенной активности фермента тирозинкиназы, которая и стимулирует опухолевый клон. В эксперименте доказано, что именно этот ген и вызывает развитие ХМЛ. Ph'-хромосому обнаруживают у 5% детей с ОЛЛ и 2% с ОМЛ. Уровень фетального гемоглобина нормальный (2%) или несколько повышен (до 7-8%). При обеих формах ХМЛ в сыворотке крови значительно повышено содержание витамина В,2. Терминальное обострение ХМЛ протекает по типу острого бластного криза с геморрагическим синдромом и интоксикацией: серо-землистой окраской кожи, генерализованной лимфаденопатией, поражением костей, гипертермией, не всегда связанной с инфекцией. Согласно А. Г. Румянцеву, властный криз у детей с ХМЛ протекает у 54% по типу миелобластного, у 26,5% больных — по типу лимфобластного, у 8,5% — смешанного. Лечение. Принципы диеты и режима, организации помощи больным те же, что и при остром лейкозе. Силенэктомия не показана. При бластных кризах лечение проводят по программам терапии острого миелоидного лейкоза. Вместе с тем установлено, что в 30% случаев при бластном кризе обнаруживают ТдТ-положительные бласты. В этих случаях сочетание преднизолона (60 мг/м2 внутрь ежедневно) и винкристина (1,5 мг/м2 внутривенно еженедельно) приводит за 4-5 нед к ремиссии. Рациональной считают следующую схему [Павлова М. Н., 1996]: 1) при наличии гиперлейкоцитоза свыше 50 тыс. в мкл назначают тиогуа-нин 60 мг/м2 внутрь ежедневно; цитозар 75 мг/м2 внутривенно сгруйно (4 дня инфузия, 4 дня перерыв). Эти два цитостатика вводят больному до снижения уровня лейкоцитоза в крови до 50 тыс. в мкл, и далее переходят к лечению литалиром; 2) литалир (гидроксикарбамид, гидреа) назначают в дозе 50 мг/кг в сутки за два приема (в капсулах) до снижения количества лейкоцитов в крови до уровня 10 тыс. в мкл, после чего подбирают индивидуальную поддерживающую дозу для больного с целью удержания количества лейкоцитов на уровне около 5000 в мкл. Параллельно со снижением количества лейкоцитов в крови на фоне приема литалира уменьшаются и размеры селезенки; 3) интерферон (интрон а-2Ь) назначают в дозе 3 млн/м2 в сутки внутримышечно. Стандартный курс лечения интерфероном 3 нед, но он может быть продолжен и далее в течение длительного времени. При этом на фоне лечения интроном получают ремиссию у 70% больных ХМЛ (у 25% она продолжается более 5 лет). Введение интрона может вызвать лихорадку, озноб и другие гриппоподобные явления: тахикардию, боли в животе, суставах, тошноту, рвоту. В этих случаях лечение временно отменяют. Схему лечения интроном ХМЛ еще отрабатывают. К сожалению, у 90% больных на фоне медикаментозной терапии и у 60% после лече- х ния интроном даже в период полной клинической ремиссии сохраняются цитогенетические изменения (Ph'-хромосома). Трансплантация костного мозга — эффективный способ лечения ХМЛ как взрослой, так и юношеской формы, но на ранних стадиях болезни или медикаментозной клинико-лабораторной ремиссии. Ювенильный вариант гораздо более устойчив к терапии, и схема его лечения не отработана. Назначают лечение по схемам ВАМП, ЦАМП и др. Поддерживающая терапия при наступлении ремиссии та же, что и при остром лейкозе. Симптоматическое лечение то же, что и при остром лейкозе. Прогноз Прогноз при ювенильном типе неблагоприятный — больные умирают в первый год лечения. При взрослом типе длительность болезни составляет несколько лет. Некоторые больные живут 10 лет и более. После успешной трансплантации костного мозга при обеих формах ХМЛ возможно выздоровление. ДЕТСКИЕ ГИСТИ0ЦИТ03Ы (ДГ). ДГ — группа конституционально обусловленных заболеваний неясной этиологии с выраженной пролиферацией клеток моноцитарно-макрофагальной системы и клеток дендритной системы. Частота Частота ДГ составляет примерно 1 случай в год на 100 тыс. детского населения в возрасте до 1 года и 2 больных в год на 1 млн детей в возрасте до 15 лет [Миллер Д. Р., 1990]. Классификация Согласно рекомендациям международной группы экспертов по ДГ (1987) следует различать следующие формы (табл. 167).
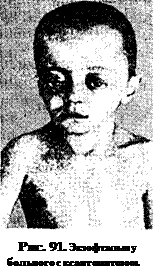
Различают три формы течения гистиоцитоза X: болезнь Абта—Леттерера— Сиве, болезнь Хенда—Шюллера—Крисчена (ксантоматоз), эозинофильная гранулема (болезнь Таратынова), которые отличаются по клинической картине и прогнозу, но описаны взаимные переходы эозинофильной гранулемы В болезнь Хенда—Шюлл ера—Крисчена, а затем в болезнь Абта—Леттерера— Сиве (в литературе 90-х годов эти три формы называют очаговый Л КГ, многоочаговый ЛКГ, диссеминированный ЛКГ). Этиология и патогенез. Этиология и патогенез ЛКГ пока не ясны. Болезнь не контагиозна; после введения материала из очагов поражения животным никаких патологических изменений у них не обнаружено. Маловероятно, что это опухоль, так как возможна спонтанная резорбция очагов поражения; более того, она типична при поражении лишь костей и длительном течении. Наиболее убедительным представляется мнение, что ЛКГ — иммунопатологический процесс с последующим цитокинопосредованным ЛКГ. Действительно, у ряда больных отмечен дефицит Т-супрессоров (CDS-лимфоциты), кортикальная атрофия в лимфатических узлах, дисплазия тимуса. У таких больных, наблюдавшихся Д. Р. Миллером (10 из 17 с ксантоматозом), достигнута полная ремиссия при лечении гормонами тимуса. При болезни Абта—Леттерера—Сиве у детей имеется глубокий комбинированный иммунодефицит с более значительным дефектом клеточного звена. В этих случаях помочь может лишь трансплантация костного мозга и иммуносупрессивная терапия. При II классе Д Г у детей выделяют обычно различные варианты герпетических вирусов (простого герпеса, цитомегалии, VI типа и др.), аденовирус. Цито-статическая и иммунодепрессивная терапия — неэффективны. Показана специфическая антивирусная терапия, иногда трансплантация костного мозга. Этиология и патогенез III класса ДГ — аналогичны лейкозам. При ЛКГ обсуждают роль вируса герпеса VI типа. Клиническая картина Болезнь Абта—Леттерера—Сиве. Развивается в большинстве случаев в раннем возрасте, преимущественно на первом году жизни. Заболевание может начинаться исподволь, с вялости, анорексии, снижения прибавки массы тела, поражения кожи (себорейный дерматит) и слизистых оболочек, субфебрилитета, а также остро, с септической лихорадки. Для периода выраженных явлений болезни характерны: 1) периодическая лихорадка септического типа; 2) изменения на коже (помимо себорейного дерматита, наблюдаются розовые папулезные высыпания в области грудины, позвоночника, покрывающиеся желтоватыми корочками, пятнисто-мелкоточечные геморрагии); 3) гепатоспленомегалия с генерализованным увеличением периферических лимфатических узлов; 4) интерстициальные поражения легких (милиарные очаги, образующие нежную сеть, распространяющуюся на оба легочных поля), иногда очаги деструкции; 5) неправильной формы деструктивные очаги в плоских костях, напоминающие при рентгенологическом исследовании географическую карту, а клинически проявляющиеся как припухлость черепа разной консистенции; 6) отиты, иногда мастоидиты, несахарное мочеизнурение, экзофтальм. При анализе периферической крови отмечают тромбоцитопению, анемию, повышенную СОЭ, лейкоцитоз с увеличением количества нейтрофилов, плазматических и ретикулярных клеток. Очень часто наслаивается вторичная инфекция и может развиться сепсис. Болезнь Хенда—Шюллера—Крисчена. Может возникнуть у детей любого возраста старше года. В типичных случаях наблюдают триаду симптомов: дефекты костей черепа, экзофтальм, несахарный диабет. Однако эта характерная триада симптомов развивается не в первые месяцы болезни и не у всех больных. Поражение костей черепа (клинически — мягкоэластическая припухлость, рентгенологически — причудливой формы очаги деструкции, напоминающие географическую карту) — наиболее постоянный симптом. Наряду с этим у больного наблюдают в различных комбинациях следующие поражения: 1) аналогичные происходящим в черепе изменения в других плоских костях, позвоночнике; 2) экзофтальм (рис. 91) и периорбитальные поражения кожи; 3) несахарное мочеизнурение; 4) задержка роста, физического и полового развития; 5) гепатомегалия, лимфаденопатия, реже гепатоспленомегалия; 6) желтоватые ксантомные очаги на коже, сочетающиеся с геморрагиями; 7) папулезные и себорейные высыпания на коже головы, спины; 8) стоматиты, рецидивирующие отиты; 9) легочные инфильтраты. В периферической крови при морфологическом исследовании отмечают лейкоцитоз, повышенную СОЭ, иногда эозинофилию, тромбоцитопению. Эозинофильная гранулема. (болезнь Таратынова) диагностируется чаще у детей дошкольного и школьного возраста и характеризуется слабостью, повышенной утомляемостью, снижением аппетита, болями в костях (нередко после травмы), гнейсом, себорейным дерматитом. Поражаются плоские (череп, таз, ребра) и трубчатые (бедро, голень, плечо) кости, а также позвоночник. На рентгенограмме очаг деструкции имеет округлую, овальную, реже неправильную форму с полициклическим фестончатым контуром без окружающего склероза. Иногда заболевание протекает бессимптомно, и очаг деструкции обнаруживают случайно, при рентгенологическом исследовании. При анализе периферической крови у больных находят увеличенную СОЭ, реже эозинофилию. Несмотря на то, что эозинофильная гранулема может самопроизвольно исчезнуть без лечения, у некоторых больных могут появиться несахарное мочеизнурение, экзофтальм, очаги деструкции в других костях, что сопровождается гепатомегалией, анемией, изменениями на коже. Наш опыт свидетельствует о возможности развития аутоиммунной гемолитической анемии у больных с эозинофильной гранулемой, причем это может быть одним из первых признаков заболевания. Гистиоцитозы II класса обычно развиваются у детей первого года жизни и начинаются с неспецифических проявлений, таких как нарушение самочувствия, плохие аппетит и прибавки массы тела, папулезно-пятнистые высыпания на коже, лихорадка. В дальнейшем выявляют панцитопению с преимущественным снижением числа эритроцитов и тромбоцитов, увеличение печени и/или селезенки, лимфатических узлов, признаки менингоэнцефалита. У некоторых больных преимущественно увеличиваются шейные лимфатические узлы и выявляют синусный гистиоцитоз. Диагноз возможен по результатам миелограммы, гистологического исследования биоптатов, где обнаруживают фагоцитирующие эритроциты — макрофаги (гистиоциты). Диагноз и дифференциальный диагноз Диагноз прежде всего основывают на характерных клинических, рентгенологических и гематологических данных. Для подтверждения диагноза гис-тиоцитозов целесообразно произвести миелограмму, биопсию кожи, лимфатического узла, пункцию пораженного участка кости черепа, биопсию очага деструкции кости. Для подтверждения диагноза выявляют гранулы Бирбека при электронной микроскопии, CD-гликопротеин в очагах поражения. Гистиоцитозы в зависимости от клинической картины заболевания дифференцируют от остеомиелита, костного туберкулеза, нейробластомы, первичных и метастатических опухолей мозга, остеокластической саркомы, фиброзной ос-теодистрофии, лимфогранулематоза, лейкоза, болезней Гоше, Нимана—Пика, портальной гипертензии с внепеченочным блоком. Лечение. Зависит от течения болезни. При остро текущих формах с генерализованным поражением внутренних органов применяют винбластин с преднизоло-ном. Винбластин вводят внутривенно 1 раз в неделю в дозе 6 мг/м2, преднизолон внутрь ежедневно по 40 мг/м2. Длительность терапии определяется ее эффективностью, но обычный курс индукции ремиссии — 7 нед, и далее переходят на поддерживающую терапию в течение 1 года: 6-меркаптопурин 50 мг/м2 + преднизолон (40 мг/м2) в 1-5-й дни недели и винбластин (6 мг/м2) в 1-й день недели. При раннем выявлении резистентности к терапии вливания винбластина сочетают с введением вепезида 150 мг/м2. Предлагают схемы комбинированной терапии с применением дексаметазона, антилимфоцитарного глобулина, вепезида и циклоспорина. Некоторые авторы используют в лечении монокло-нальные'антитела к CDl-антигену. Дата добавления: 2014-10-03 | Просмотры: 1600 | Нарушение авторских прав |