|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Генез наиболее важных синдромов при ХПН. 8 страница
— врожденную трансиммунную (транзиторная тромбоцитопения новорожденных от матерей, больных идиопатической тромбоцитопеничес-кой пурпурой, красной волчанкой). Вторичные (симптоматические) тромбоцитопении у детей развиваются чаще первичных и могут наблюдаться: — в острый период инфекционных заболеваний (особенно часто при перинатальных вирусных инфекциях); — при аллергических реакциях и болезнях, протекающих с гиперреактивностью немедленного типа; — коллагенозах и других аутоиммунных расстройствах; — ДВС-синдроме; — злокачественных заболеваниях системы крови (лейкоз, гипопластичес-кие и витамин В12-дефицитные анемии); — болезнях, сопровождающихся спленомегалией и дисспленизмом (портальная гипертензия при циррозах печени и др.); — врожденных аномалиях сосудов (гемангиомы) и обмена веществ (Гоше, Ниманна—Пика и др.).
Идиопатическая тромбоцитопеническая пурпура (ИТП). ИТП — первичный геморрагический диатез, обусловленный количественной и качественной недостаточностью тромбоцитарного звена гемостаза. Характерными признаками болезни являются нарушения в сосудисто-тромбо-цитарном звене гемостаза — пурпура (кровоизлияния в толще кожи и слизистых оболочках) и кровоточивость слизистых оболочек, низкое количество тромбоцитов в периферической крови, нормальное или повышенное количество мегакариоцитов в костном мозге, отсутствие спленомегалии и системных заболеваний, течение которых может осложниться тромбоцитопенией. Этиология и патогенез
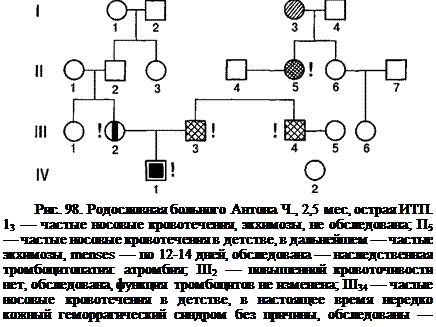
ИТП — заболевание с наследственным предрасположением, заключающемся в наличии у больных наследственной тромбоцитопатии (рис. 98), в связи с чем перенесенные вирусные инфекции (ОРВИ, корь, краснуха и др.), профилактические прививки, физические и психические травмы и другие внешние факторы могут привести к нарушению антигенпрезентующей функции макрофагов и далее возникновению иммунопатологического процесса — пролиферации сенсибилизированных к аутотромбоцитам лимфоцитов, синтезу антитромбоцитарных аутоантител. По современным представлениям, к ИТП всегда приводит иммунопатологический процесс, поэтому выделение иммунных и неиммунных форм болезни нерационально. Семейные особенности иммунологической реактивности, иммунорегуляции могут быть также фактором наследственного предрасположения к ИТП (наряду, конечно, с наличием наследственной тромбоцитопатии). Антитромбоцитарные аутоантитела при ИТП направлены к различным гликопротеинам (ГП) мембраны тромбоцитов; ITIV (обычно у таких детей развиваются спонтанная ремиссия), ГП lb, ГП lib — Ша (высокий титр типичен для хронической ИТП). У части больных ИТП снижено количество Т-лимфоцитов супрессоров и естественных киллеров. Формальный генез тромбоцитопении при ИТП не вызывает сомнений — повышенная деструкция тромбоцитов в селезенке, которая одновременно является местом синтеза антитромбоцитарных антител. Тромбоцитопоэз при ИТП повышен, на что указывает большое количество мегатромбоцитов в крови. Кровоточивость у больных ИТП обусловлена количественной (тромбоци-топения) и качественной (тромбоцитопатия) неполноценностью тромбоцитарного звена гемостаза. Сосудистый эндотелий, лишенный ангиотрофической функции тромбоцитов, подвергается дистрофии, что приводит к повышению проницаемости сосудов, спонтанным геморрагиям. Нарушения коагуляцион-ного звена гемостаза у больных ИТП (снижение темпов тромбопластинообра-зования, повышение фибринолиза) вторичны по отношению к недостаточности тромбоцитарного звена. Тромбоцитопатию у детей, больных ИТП, отмечают во все периоды заболевания, в том числе и после спленэктомии при нормальном количестве тромбоцитов в периферической крови. Аналогичную ситуацию отмечают при анемии Минковского—Шоффара, когда после спленэктомии анемия исчезает, а микросфероцитоз сохраняется.
Классификация По течению выделяют острые (продолжающиеся менее 6 мес) и хронические формы ИТП; последние подразделяют на варианты: а) с редкими рецидивами; б) с частыми рецидивами; в) непрерывно рецидивирующие. По периоду болезни различают обострение (криз), клиническую ремиссию (отсутствие кровоточивости при сохраняющейся тромбоцитопении) и клинике-гематологическую ремиссию.
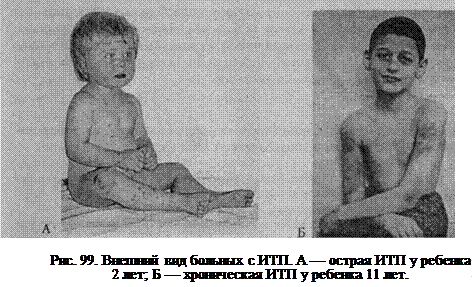
По клинической картине различаются «сухие»' (имеется только кожный геморрагический синдром) и «влажные» (пурпура в сочетании с кровотечениями) пурпуры. Клиническая картина. ИТП в большинстве случаев развивается в детском возрасте, главным образом у дошкольников. До начала периода полового созревания среди больных ИТП мальчики и девочки встречаются одинаково часто, но среди старших школьников девочек уже в 2 раза больше. В раннем и дошкольном возрасте ИТП чаще развивается через 2-4 нед после перенесенных вирусных инфекций: появляются петехиально-пятнистые кожные геморрагии, кровоизлияния в слизистые оболочки, кровотечения. Характерными чертами пурпуры у детей являются: 1) полихромность (одновременно на коже можно обнаружить геморрагии разной окраски — от красновато-синеватых до зеленых и желтых); 2) полиморфность (наряду с разной величины экхимозами имеются пете-хии); 3) несимметричность; 4) спонтанность возникновения, преимущественно по ночам (рис. 99).
Умеренная спленомегалия (нижний край селезенки на 1-2 см выступает из-под реберной дуги) может быть обнаружена у 10% детей, больных ИТП. Температура тела при отсутствии сопутствующих заболеваний и постгеморрагической анемии стойко нормальная. Диагноз. Наиболее характерными отклонениями от нормы при лабораторном обследовании больных ИТП являются тромбоцитопения (норма тромбоцитов в периферической крови 150-400 х 109/л), увеличение времени кровотечения после стандартной травмы (по Дюку, но лучше по Айви или Борхгревину—Ваалеру), положительные пробы на резистентность капилляров (жгута, щипка и др.), увеличение количества «недеятельных» мегакариоцитов в костном мозге. Миело-грамма обязательна с целью дифференциального диагноза. При дифференциальном диагнозе следует иметь в виду, что стойкая гипоплазия мегакариоцитарного ростка, изменения миелоидного и эритроидного ростков костного мозга при отсутствии постгеморрагической анемии нехарактерны для ИТП, и наличие их требует поисков других причин тромбоцитопении. Лихорадка, увеличение лимфатических узлов, спленомегалия, обнаружение выраженных сдвигов в лейкоцитарной формуле, лейкоцитоза, повышенная СОЭ, значительная диспротеинемия — все это требует исключения инфекционных и других перечисленных выше заболеваний и нетипично для ИТП. Особенно важно дифференцировать ИТП с злокачественными ге-мопатиями (лейкозы и др.), иммунопатологическими болезнями (системная красная волчанка и др.). Врожденная изоиммунная тромбоцитопеническая пурпура. Обычно возникает при наличии у плода тромбоцитарного антигена PLAI и отсутствии его у матери (в популяции таких лиц 2-5%), что и приводит к сенсибилизации матери, синтезу ею антитромбоцитарных антител, которые, проникая через плаценту, вызывают тромбоцитолиз у плода. Заболевание диагностируют у 1 на 5000-10 000 новорожденных; характеризуется появлением в первые часы жизни петехиальных и мелкопятнистых геморрагии на коже. При тяжелом течении (позднее появление геморрагического синдрома) возможны кровоизлияния в слизистых оболочках, мелена, носовые, пупочные, легочные кровотечения, внутричерепные геморрагии (у 10-12% больных). Типична умеренная спленомегалия. Диагноз основан на клинико-лабораторных данных, положительной реакции тромбоагглютинации кровяных пластинок ребенка в сыворотке крови матери. Тромбоцитопения держится от 2 до 12 нед, а иногда и дольше, хотя нарастание геморрагического синдрома купируется при рациональной терапии в первые дни жизни. Врожденная трансиммунная тромбоцитопения. В период новорожден ности наблюдается у 30-50% детей от матерей, больных идиопатической тром-боцитопенической пурпурой. В половине случаев не сопровождается геморрагическими расстройствами. Генез тромбоцитопении связан с проникновением от матери к плоду антитромбоцитарных антител и сенсибилизированных к аутотромбоцитам материнских лимфоцитов. Клинические проявления развиваются обычно в первые дни жизни: петехии, небольшие экхимозы на спине, груди, конечностях, реже кровоизлияния на слизистых оболочках, мелена, носовые кровотечения. Как правило, кровоточивость нетяжелая, но описаны случаи внутричерепных геморрагии. Диагноз основан на анамнестических и кли-нико-лабораторных данных с обнаружением у матери антитромбоцитарных аутоантител, а у матери и ребенка — сенсибилизированных к аутотромбоцитам лимфоцитов. Выздоровление наступает через 5-12 нед. Переход в идиопати-ческую тромбоцитопеническую пурпуру наблюдается в 1-3% случаев. Наследственные тромбоцитопенические пурпуры могут быть следствием недостаточного образования тромбоцитов или повышенного их разрушения. Гипопластические тромбоцитопенические пурпуры (с амегакариоцитозом или мегакариоцитарной гипоплазией), как правило, сочетаются с другими пороками развития, особенно часто с аплазией лучевых костей. Кровоточивость (пурпура, носовые, кишечные и другие кровотечения) и тромбоцитопения в типичных случаях появляются в первые дни жизни, реже позже. Наследование аутосомное, рецессивное. Прогноз плохой: более половины больных умирает в возрасте до года. Гипоплазия мегакариоцитарного ростка может быть симптомом гипопластических анемий, лейкоза, хромосомных аномалий (трисомий по 13-15-й, 18-й парам хромосом). Наследственная тромбоцитопеническая пурпура может быть и результатом дефицита синтеза тромбоцитопоэтина (доминантный тип наследования), и в таких случаях назначение плазмы даже внутрь приводит к временной нормализации количества тромбоцитов. Наследственные тромбоцитолитические пурпуры. Бывают микроци-тарные, макроцитарные и нормоцитарные. Из этой группы наиболее изучены синдром Вискотта—Олдрича (см. главу23), геморрагическая тромбоцитоди-строфия (синдром Бернара—Сулье) и аномалия Мея—Хегглина. При синдроме Бернара—Сулье, наследуемом по рецессивно-аутосомному типу, кровоточивость чаще тяжелая: в первые месяцы жизни появляется пурпура, кровотечения. Тромбоцитопения либо появляется, либо значительно нарастает в момент кровотечения. Тромбоциты большие (80% их имеют диаметр более 4 мкм, большой голубой гиаломер), не реагируют на такие агрегирующие агенты, как ристоцетин, бычий фибриноген. Сущность аномалии тромбоцитов — отсутствие в оболочке клеток гликопротеина lb (гликокаль-цина), содержащего рецепторы для фактора Виллебранда, что и определяет низкую адгезивную способность тромбоцитов и дефект агрегации с ристоцетином. Кровоточивость при синдроме Мая—Хегглина нетяжелая, возникает в дошкольном и школьном возрасте. Наследование аномалии аутосомное, доминантное. Тромбоцитопения сочетается с большими размерами кровяных пластинок и наличием телец Доле в нейтрофилах и моноцитах. Лечение. Зависит от генеза тромбоцитопении. Новорожденных с изоиммунными и трансиммунными пурпурами в течение 2 нед кормят донорским грудным молоком, а затем прикладывают к груди матери (с контролем числа тромбоцитов в периферической крови ребенка). При других тромбоцитопенических пурпурах детей кормят обычно, в соответствии с возрастом. Режимные ограничения, как правило, необходимы лишь в период геморрагического криза. При любой тромбоцитопенической пурпуре (если исключен ДВС-синдром) показано назначение внутрь е-аминокапроновой кислоты (0,05-0,1 г/кг 4 раза в сутки) и других препаратов, улучшающих адгезивно-агрегационную активность тромбоцитов (адроксон, этамзилат-дицинон, пантотенат кальция, хлорофиллин натрия, АТФ внутримышечно в сочетании с препаратами магния внутрь, фитотерапия). В период геморрагического криза е-аминокапро-новую кислоту необходимо 1-2 раза в день вводить внутривенно капельно. Иммуноглобулин внутривенно капельно вводят в дозе 0,5 г/кг массы тела (10 мл/кг 5% раствора) ежедневно в течение 4 дней, что приводит к подъему количества тромбоцитов к концу курса лечения у 2/3~3/, больных ИТП, хотя лишь у 25-30% больных достигается стойкая клинико-гематологическая ремиссия. Лечение внутривенным иммуноглобулином показано и при трансиммунной и изоиммунных тромбоцитопенических пурпурах новорожденных. Однако это достаточно дорогой метод и обычно все же у детей с ИТП лечение начинают с применения преднизолона. Показаниями к назначению глюкокортикоидов детям, больным ИТП, являются: генерализованный кожный геморрагический синдром при количестве тромбоцитов менее 20 х 109/л, сочетающийся с кровоточивостью слизистых оболочек, кровоизлияния в склеру и сетчатку глаза, влажная пурпура, осложнившаяся постгеморрагической анемией, кровоизлияния во внутренние органы. Преднизолон назначают на 2-3 нед в дозе 2 мг/кг в сутки с дальнейшим снижением дозы и отменой препарата. Длительное, многомесячное лечение глюкокортикоидами неэффективно и приводит к ряду осложнений. Антирезусный иммуноглобулин (анти-D-IgG), вводимый резус-положительным больным внутривенно в дозе 25-75 мкг/кг в течение 2-5 дней у половины больных с хронической ИТП, способствует повышению количества тромбоцитов. У спленэктомированных неэффективен. Интерферон-а генноинженерный препарат (Реаферон, Интрон А, Реафе-рон-А), начали использовать для лечения больных ИТП с 1988 г. Согласно Е. К. Донюш и соавт. (1997), из 40 детей с хронической ИТП гематологический ответ был достигнут у 72,5% больных (у 15% полный, то есть нормализация количества тромбоцитов). Детям до 5 лет доза препарата составляла 500 000 МЕ/сутки, 5-12 лет — 1 млн МЕ/сутки и старше 12 лет — 2 млн МЕ/сутки. Препарат вводили подкожно или внутримышечно 3 раза в неделю в течение 3 мес. Хотя результаты лечения следует расценивать лишь через 6-7 нед от его начала, у большинства больных гематологический эффект отмечался уже через 2 нед. Трансфузии тромбоцитной массы — не эффективны, и к ним прибегают очень редко, лишь при жизнеугрожающих кровоизлияниях (например, в головной мозг), количество тромбоцитов в периферической крови при этом не увеличивается, и возможен только местный гемостатический эффект. Показания к спленэктомии у больных ИТП: влажная пурпура, продолжающаяся более 6 мес и требующая назначения повторных курсов глюкокортикоидов; острая пурпура при наличии тяжелой кровоточивости, не купирующейся на фоне современной комплексной терапии; подозрение на кровоизлияние в мозг. По суммарным данным литературы, у 85% детей, больных ИТП, спленэк-томия приводит к клинико-лабораторной ремиссии либо к значительному уменьшению кровоточивости. Самой частой причиной рецидива болезни после спленэктомии является наличие у больных добавочных селезенок, не удаленных во время операции, что можно определить по отсутствию эритроцитов с тельцами Ховеля—Жоли, которые обязательно появляются у спленэктоми-рованных. В случае неэффективности спленэктомии, вновь кратковременно применяют глюкокортикоиды, а если и это не приводит к увеличению числа тромбоцитов и купированию геморрагии, назначают винкристин (1,5 мг/м2) внутривенно 1 раз в неделю. Обычно эффект наступает после 2-4 введений. Более длительно иммунодепрессоры давать нецелесообразно. Используют после спленэктомии и аттенуированный андроген даназол (20 мг/кг в сутки, не более 800 мг, разделенных на две дозы) в течение 1-12 мес. Спленэктомия нежелательна у детей в возрасте до 5 лет (особенно до года), так как риск сепсиса в этом случае около 1-2%. Учитывая, что спленэктоми-рованные больные особенно чувствительны к пневмококковой инфекции (лишь в первый год после операции), рекомендуют до спленэктомии провести иммунизацию пневмококковой вакциной, либо в течение полугода после операции 1 раз в месяц вводить бициллин-5. Диспансерное наблюдение. При острой ИТП проводят в течение 5 лет, при хронической — до перехода ребенка во взрослую поликлинику. Прививки на фоне антигистаминных средств возможны лишь через 1 год после острого периода. Противопоказаны прививки живыми вирусными вакцинами. В течение 3-5 лет нецелесообразна смена климата. Диета обычная. Анализы крови с подсчетом числа тромбоцитов в первые 3 мес после выписки надо делать 1 раз в 2 нед, далее в течение 9 мес ежемесячно и в дальнейшем 1 раз в 2 мес, а также после каждой перенесенной инфекции. При лечении детей следует избегать ацетилсалициловой кислоты, анальгина, карбенициллина, нитрофурановых препаратов, УВЧ и УФО.В течение 3-6 мес после выписки целесообразно назначить кровоостанавливающий сбор трав в сочетании с чередующимися 2-недельными курсами препатаров, стимулирующих адгезивно-агрегационную функцию тромбоцитов, желчегонными средствами (прием аллохола в течение 1 мес, затем перерыв на 1 мес и снова прием этого препарата). Далее упомянутую терапию назначают курсами по 2 мес 2-3 раза в год. В кровоостанавливающий сбор трав входят тысячелистник, пастушья сумка, крапива двудомная, зайцегуб опьяняющий, зверобой, земляника лесная (растения и ягоды), водяной перец, кукурузные рыльца, шиповник. Все растения смешивают в равных частях, заливают 1 столовую ложку сбора стаканом кипятка и настаивают 10-15 мин. Пить по 1 стакану в сутки в 2-3 приема в течение 1 мес. Затем делают перерыв на 1 мес и вновь проводят месячный курс. Прогноз При современных методах лечения для жизни — благоприятный. Летальность не выше 2-3%. По общепринятому мнению, 75% больных с острой ИТП выздоравливают спонтанно, без всякого лечения в течение 6 мес. Частота перехода острой ИТП в хроническую составляет примерно 10%.
Тромбоцитопатии. Тромбоцитопатии — расстройства гемостаза, обусловленные качественной неполноценностью кровяных пластинок при нормальном их количестве. Различают наследственные и приобретенные тромбоцитопатии. Среди первичных наследственных тромбоцитарных дисфункций наиболее часто встречаются атромбия, тромбоцитопатия с дефектом реакции освобождения, тромбастения. Вторичные наследственные тромбоцитопатии типичны при болезни Виллебранда, афибриногенемии, альбинизме (синдром Херманско-го—Пудлака), синдроме гиперэластичной кожи (Элерса—Данлоса) и Марфа-на, многих аномалиях обмена. Приобретенные тромбоцитопатии с геморрагическим синдромом или без него характерны для многих болезней крови (лейкозы, гипопластические и мегалобластические анемии), уремии, ДВС-синдрома, иммунопатологических болезней (геморрагический васкулит, красная волчанка, диффузный гломерулонефрит и др.), лучевой болезни, лекарственной болезни при приеме салицилатов, ксантинов, карбенициллина, нейроциркуляторных дисфункций. Распространение даже первичных наследственных тромбоцитопатии не установлено, но несомненно, что это самая частая генетически обусловленная патология системы гемостаза. В большинстве случаев так называемой семейной кровоточивости неясного генеза можно подозревать наследственные тромбоцитопатии. Частота наследственных тромбоцитопатии в популяции достигает 5% и более. Патогенез. В основе дефекта функциональной активности тромбоцитов при тромбас-тении (описана в 1918 г. швейцарским педиатром Евгением Гланцманном и потому названа его именем) лежит отсутствие комплекса гликопротеинов ПЬ/ Ша на их мембране, а отсюда неспособность тромбоцитов связывать фибриноген, агрегировать друг с другом, вызывать ретракцию кровяного сгустка. Комбинация гликопротеинов lib/Ша является специфическим для тромбоцитов и мегакариоцитов интегрином — комплексом, опосредующим различные внеклеточные сигналы к цитоскелету тромбоцитов. Классификация наследственных тромбоцитопатий [R. Colman, 1993] I. Дефект взаимодействия тромбоцит—агрегант (дефект рецептора): — селективные дефекты адреналиновых, коллагеновых, тромбоксано-вых/ эндопероксидных рецепторов. II. Дефект взаимодействия тромбоцит—сосудистая стенка: — болезнь Виллебранда (дефицит или дефектность ФВ); — синдром Бернара—Сулье (дефицит ГП lb). III. Дефект межтромбоцитарного взаимодействия: — врожденная афибриногенемия (дефицит фибриногена); — тромбастения Гланцманна (дефицит или дефект ГП ИЬ/Ша). IV. Нарушения тромбоцитарной секреции: 1) дефицит пула хранения: — дефицит 8-гранул; — дефицит а-гранул; — дефицит а8-гранул. 2) нарушения обмена арахидоновой кислоты: — нарушения высвобождения арахидоновой кислоты; — дефицит циклооксигеназы; — дефицит тромбоксансинтетазы. 3) первичный дефект секреции с нормальным пулом хранения и нор- — дефект мобилизации кальция; — дефект метаболизма фосфатидилинозитола; — дефект фосфорилирования белков.
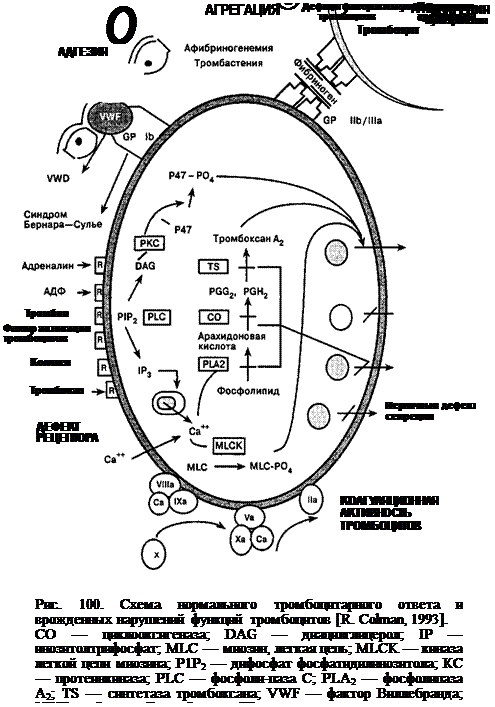
V. Нарушения взаимодействия тромбоцит—фактор свертывания. VI. Сочетанные врожденные нарушения — аномалия Мея—Хегглина, болезнь Дауна, синдромы мезенхимальной дисплазии (Марфана, Элерса— Данлоса), TAR-синдром. Достижения последнего десятилетия в области молекулярной биологии, биохимии мембран, механизмов трансмембранной передачи сигналов к внутриклеточному скелету, а также участие вторых посредников (аденилатцик-лазного — тормозного и Са-инозитолового — стимулирующего) в реализации клеточных эффектов способствовали пониманию путей взаимодействия клеток крови, эндотелиоцитов с биологически активными веществами в ходе воспалительного процесса, репарации, гемостаза, аллергии. Наследственные дефекты мембран — причина неспособности тромбоцитов реагировать на тромбоксан при атромбии или связывать фактор Виллебранда, адгезировать (прилипать) к чужеродным поверхностям, коллагену при аномалии Бернара—Сулье. При разных вариантах наследственных тромбо-цитопатий с дефектом реакции освобождения выявлены дефициты циклоок-сигеназы, тромбоксан-синтетазы и др. Кроме того, при некоторых наследственных тромбоцитопатиях обнаружен дефицит плотных гранул (болезнь Херманского—Пудлака, синдром Ландольта), дефицит белковых гранул (синдром серых тромбоцитов) или их компонентов, лизосом. В генезе повышенной кровоточивости при всех вариантах тромбоцитопатии основное значение имеет дефект образования первичной гемостатической пробки и взаимодействия как самих тромбоцитов друг с другом, так и тромбоцитарного и плазменного звеньев гемостаза. Современные представления о рецепторном аппарате тромбоцитов и врожденных тромбоцитопатиях представлены на рис. 100, классификация наследственных тромбоцитопатии [Colman R., 1993] — см. стр. 336. Однако практическое использование данной классификации достаточно сложно и возможно лишь в высокоспециализированных клиниках, поэтому для практических целей можно использовать следующую упрощенную классификацию (табл. 173). Клиническая картина
месте сдавления резинками или при энергичном давлении на конечность; периодические необильные носовые кровотечения, семейные длительные менструации у женщин и др.) до обильных носовых, маточных, желудочно-кишечных кровотечений, распространенной кожной пурпуры. Нередко у таких больных небольшие хирургические вмешательства (экстракция зуба, адено-томия, тонзиллэктомия и др.) вызывают обильные и длительные кровотечения, приводящие к анемии. Кожный геморрагический синдром может быть в виде петехий, экхимозов, реже гематом. Часто минимальная кровоточивость настолько распространена среди родственников, что это объясняют семейной слабостью сосудов, семейной чувствительностью и т. д. Именно у больных с наследственными тромбоцитопатиями развивается обычно кровоточивость как осложнение после приема лекарственных препаратов, которые у миллионов людей этой реакции не вызывают. У них же часты носовые кровотечения при инфекциях. Длительные торпидные к обычной терапии гематурии тоже могут быть проявлением тромбоцитопатии (все же у таких больных всегда либо в анамнезе, либо даже в момент обследования можно обнаружить и другие проявления повышенной кровоточивости). Время появления первых признаков повышенной кровоточивости может быть самым различным, но чаще все-таки это ранний или дошкольный возраст. Весной и зимой кровоточивость более выраженная. С возрастом нередко интенсивность геморрагического синдрома стихает. Наиболее стойкий и тяжелый геморрагический синдром из перечисленных наследственных тромбоцитопатии отмечается при тромбастении. Частота различных симптомов повышенной кровоточивости при различных тромбоцитопатиях представлена на рис. 101.
а ь с d
—пурпура |=j —носовые Xffl\ —гематурия t=J кровотечения У//Л -метроррагия шВ _ выражанные геморрагии после операций, травм
Рис. 101. Характер повышенной кровоточивости при наследственных тромбоцитопатиях (Е. Д. Болотина). а — тромбастения; b — атромбия; с — дефект реакции освобождения; d — болезнь Виллебранда. Диагноз и дифференциальный диагноз. Заболевание можно заподозрить уже на основании анамнеза. Обязательным является составление родословной с тщательным сбором сведений о минимальной кровоточивости у родственников. Эндотелиальные пробы на резистентность капилляров (манжеточная, жгута, баночная), как правило, положительные. Может быть увеличена длительность кровотечения, но при определении по Дюку она чаще нормальная. Количество тромбоцитов и показатели свертывающей системы крови без отклонений от нормы (во всяком случае, при определении традиционными методами изучения коагулограм-мы). Окончательный диагноз возможен лишь при лабораторном исследовании свойств тромбоцитов: их адгезивной способности к стеклу и коллагену (снижены лишь при болезнях Виллебранда и Бернара—Сулье), ретенции их в ранке, агрегационной активности с разными дозами АДФ, коллагеном, ристоце-тином, оценке реакций освобождения. При этом обследование следует проводить в динамике (для исключения случайных изменений свойств тромбоцитов) и обязательно не только у ребенка, но и его родителей, а также родственников с повышенной кровоточивостью.
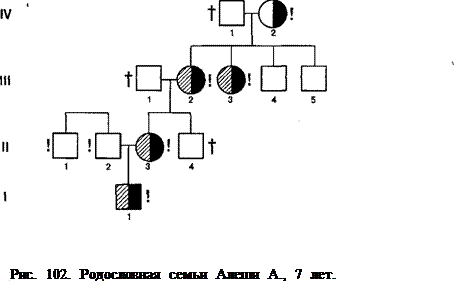
Атромбию, тромбоцитопатию с дефектом реакций освобождения наследуют по аутосомно-доминантному типу (рис.102, 103), поэтому у одного из родителей больного дефект свойств тромбоцитов находят обязательно. Тромба-стению нередко наследуют аутосомно-рецессивно, в связи с чем выявление гетерозиготного носителя среди родителей может быть затруднено (рис.104). В то же время, есть семьи и с доминантным наследованием тромбастении. tD3 74 80
DO 50 40 46
8 9 10 11 !«ООО 49 57
DO 4 ]J5 6 7 8 9 110 □•olOniiAoс 25 34
Рис. 103. Родословная семьи Миши М., 3,5 лет. Наследственная тромбоцитопатия с дефектом реакции освобождения. Ill ОтООтП
Dr#! 27 27
I _ 7 2,5 Рис. 104. Родословная семьи Лены Р., 2,5 лет. Тромбастения. Дата добавления: 2014-10-03 | Просмотры: 1773 | Нарушение авторских прав |


 54
54