|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Генез наиболее важных синдромов при ХПН. 6 страница
При выявлении дефицита С08-лимфоцитов показано лечение гормонами тимуса. При эозинофильной гранулеме и болезни Хенда—Шюллера—Крисчена, протекающей лишь с поражением костей и/или несахарным диабетом, необходимости в таком массивном лечении, как правило, нет. В то же время, изолированное применение глюкокортикоидов при любых формах гистиоцитоза X считают неадекватной терапией. Эти препараты целесообразно назначать только с цитостатиками. При очаговых поражениях костей и на область турецкого седла при несахарном диабете применяют лучевую терапию. Несахарное мочеизнурение — показание к лечению адиурекрином или десмопрессином. При ограниченных кожных поражениях применяют мази с глюкокортикоидами, дексаметазоном, мустардом. При II типе ДГ положительный эффект оказывает специфическая проти-воинфекционная, то есть противовирусная, терапия. Прогноз Определяется степенью генерализации процесса. Считают, что при отсутствии поражения внутренних органов даже при наличии генерализованного поражения костей прогноз благоприятный. При эозинофильной гранулеме прогноз, как правило, тоже благоприятный, а при болезни Абта—Леттерера— Сиве все же чаще неблагоприятный. Общий процент выздоровлений при всех формах гистиоцитоза X — 70%.
ЛИМФОГРАНУЛЕМАТОЗ (ЛГМ). Лимфогранулематоз (болезнь Ходжкина) — злокачественная лим-фома, был впервые описан английским врачом Т. Ходжкином в 1832 г. Частота ЛГМ составляет примерно 10% всех опухолевых заболеваний у детей. У мальчиков ЛГМ встречается примерно вдвое чаще, чем у девочек. Заболеваемость ЛГМ составляет примерно 1 случай в год на 100 ООО детского насе-- ления. Обычно заболевают дети старше 4-5 лет. Есть неясные указания на большую частоту развития ЛГМ у тонзиллэктомированных детей. Этиология и патогенез Причина ЛГМ неизвестна. Вирусная гипотеза основана на обнаружении ДНК вируса Эпштейна—Барр в очагах поражения и антител к этому вирусу в крови у части детей с ЛГМ. Однако большинство авторов считает это скорее маркером нарастающего иммунодефицитного состояния, характерного для ЛГМ, проявляющегося в прогрессирующем уменьшении абсолютного количества Т-лимфоцитов, угнетении их функциональной активности. Больные ЛГМ становятся высокочувствительными к вирусам, микробным и грибковым инфекциям. В конечных стадиях ЛГМ у взрослых состояние больных и иммунный статус напоминают болезнь «трансплантат против хозяина». У детей угнетение Т-иммунитета бывает гораздо реже и менее глубокое. При ЛГМ не обнаружено каких-либо постоянных хромосомных нарушений (за исключением гиперплоидности клеток Рида—Березовского—Штернберга). Опухолевая природа ЛГМ имеет наибольшее количество сторонников, основывающих свое мнение на неуклонном прогрессировании болезни без лечения, склонности к инфильтративному росту в терминальной стадии болезни. Полагают, что при ЛГМ имеет место уницентрический (клональный) генез процесса с происхождением из клеток-предшественников моноцитарно-мак-рофагального ряда. Классификация. В 1989 г. Международный многопрофильный комитет модифицировал предшествующие классификации ЛГ следующим образом (табл. 168). При верификации стадийности ЛГМ принимают во внимание результаты тщательного физикального исследования, рентгенологического обследования (рентгенограмма грудной клетки, компьютерная томография грудной клетки, живота и таза), полного клинического анализа крови, двусторонней тре-панобиопсии, гистологического изучения биоптата лимфоузла, а по показаниям — лимфангиографии нижних конечностей, лапаротомии. Гистологические варианты ЛГМ: 1. Нодулярный склероз — наиболее распространенный вариант, встречается у 50% детей с ЛГМ, имеет хороший прогноз. При окраске гематоксилин-эозином обнаруживают эозинофильные коллагеновые волокна различной ширины, разделяющие лимфоузлы на отдельные голубоватые лимфоидные узелки (нодули). При II степени склероза встречаются
Таблица 168 Классификация лимфогранулематоза Котсволд.
участки, обедненные лимфоцитами или с большим количеством клеток Рида—Березовского—Штернберга. Иногда обнаруживают очаги склероза. При первой стадии количество клеток Рида—Березовского-Штернберга невелико. 2. Вариант с лимфоидным преобладанием — пораженная ткань состоит из мелких лимфоцитов и доброкачественных эпителиоидных гистиоцитов. Клетки Рида—Березовского—Штернберга встречаются редко. Прогноз — неплохой, если нет нодулярного варианта. 3. Лимфоидпое истощение — вариант, при котором в биоптате выявляют очаги некрозов, много клеток Рида—Березовского—Штернберга и очень малое количество лимфоцитов. Обычно этот вариант связан с IV стадией ЛГМ с инфильтрацией костного мозга, истощающей лихорадкой. 4. Смешанно-клеточный вариант (второй по частоте встречаемости у детей с ЛГМ) характеризуется наличием большого количества типичных клеток Рида—Березовского—Штернберга, окруженных реактивными гистиоцитами, мелкими лимфоцитами, нейтрофилами, эозинофилами, плазматическими клетками и мелкими очагами некроза. Заболевание может протекать как фокальное или частичное поражение лимфоузлов, но может характеризоваться быстрым прогрессированием. Клетки Рида—Березовского—Штернберга описаны С. Я. Березовским в 1890 г., К. Штернбергом в 1898 г. и наиболее тщательно Д. Ридом в 1902 г. и представляют собой гигантские клетки (диаметр 30-80 мкм) с двух- и более дольчатым ядром и огромными эозинофильными, похожими на включения, ядрышками. Часто их описывают как многоядерные клетки. Классические клетки Рида—Березовского—Штернберга имеют зеркально симметричные ядра, выглядящие как глаза совы; цитоплазма клетки бледная (светло-серо-
I
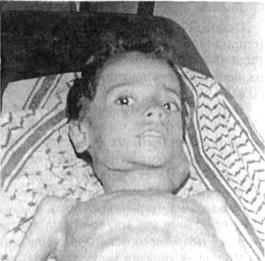
Рис. 92. Увеличение шейных лимфатических узлов у больного ЛГМ. больных детей с ЛГМ процесс начинается с увеличения лимфоузлов средостения. Реже первичный процесс начинается с увеличения лимфоузлов подмышечной области, подчелюстных, брюшной полости. Лишь у 10% больных при первом обращении обнаруживают поражения лимфоузлов по обе стороны диафрагмы. При фокальном увеличении лимфоузлов лишь шейной области больные могут не предъявлять каких-либо жалоб, у них нет признаков интоксикации. По мере распространения процесса нарастают слабость, повышенная утомляемость, снижается аппетит, ребенок худеет, появляются необъяснимая лихорадка, ночная потливость. Особенно типичны для ЛГМ волнообразная лихорадка, ночные поты, кожный зуд. Лихорадка, профузные ночные поты, похудание (потеря 10% массы тела), зуд кожи, увеличение размеров печени с нарушением ее функций — признаки интоксикации, то есть В-сим-птомы. Быстрое развитие симптомов интоксикации характерно для поражения брюшных лимфоузлов, когда рано вовлекаются в процесс паренхиматозные органы. При прогрессировании ЛГМ могут поражаться также легкие, плевра, желудочно-кишечный тракт, костный мозг, скелет, головной и спинной мозг (IV стадия болезни). Прогрессирующий иммунодефицит обуславливает предрасположенность больных ЛГМ к инфекционным заболеваниям. Особенно тяжело при ЛГМ протекают корь, инфекции, вызванные герпетическими вирусами (ветряная оспа, герпес-зостер, цитомегалия и др.) Диагноз. Основой диагностики должно быть гистологическое исследование взятого при биопсии пораженного лимфоузла и обнаружение в материале клеток Рида—Березовского—Штернберга. Игловая пункция лимфоузла нежелательна, ибо не дает основания для решения вопроса о гистологическом варианте ЛГМ. При анализе периферической крови для ЛГМ типично резкое увеличение СОЭ и нейтрофилез, нарастающая лимфоцитопения. Общее количество лейкоцитов может быть как нормальным, так и слегка увеличенным или сниженным. По мере прогрессирования болезни появляются анемия, тромбоцитопе-ния. На ранних стадиях болезни в миелограмме выявляют раздражение гранулоцитопоэза, эритропоэза, мегакариоцитопоэза, эозинофильную или ре-тикулоплазмоцитарную реакцию, но по мере прогрессирования ЛГМ нарастают угнетение как миело-, так и эритро-мегакариоцитопоэза, появляются клетки Рида—Березовского—Штернберга. В терминальных стадиях возможна тотальная аплазия костного мозга, частично обусловленная и комплексной цитостатической терапией. К числу обязательных методов обследования относят также: рентгенографию грудной клетки в двух проекциях, сцинтиграфию печени и селезенки, нижнюю лимфосцинтиграфию. Лапаротомия для уточнения стадии показана больным ЛГМ с I, II, ША стадиями, которым в качестве терапии планируют облучение, ибо у 25% таких больных имеются оккультные поражения лимфоузлов брюшной полости. Если при лапаротомии будут обнаружены в значительном количестве лимфоузлы в воротах селезенки (4 лимфоузла и более) или увеличенные парааортальные лимфоузлы, то преимущество для таких больных будет иметь комбинированная терапия. Лапаротомия не показана больным ЛГМ с ШВ или IV стадией, которым планируется химиотерапия, а также больным с большой опухолью средостения (преимущество имеет комбинированная терапия), при изолированном поражении шейных лимфоузлов или больным, не имеющим большого поражения средостения с гистологическим вариантом нодулярного склероза (достаточно субтотального облучения пораженных лимфоузлов). Дифференциальный диагноз. При шейной форме ЛГМ исключают вульгарный и туберкулезный лимфаденит. В таких случаях нередко обнаруживают очаги хронической инфекции в полости рта (периодонтит, хронический тонзиллит и др.), носоглотки (адено-идиты и др.), придаточных пазух. Могут быть выражены симптомы интоксикации, воспалительные изменения в крови, пальпируют размягчение лимфоузла в центре. Для туберкулеза характерно наличие очага поражения в легких. Кроме того, имеют в виду и болезнь Брилла—Симмерса, инфекционный мононуклеоз, лейкоз. При поражении средостения следует дифференцировать с туберкулезом, саркоидозом, опухолями вилочковой железы, неходжкинскими лимфомами, дермоидными кистами. При внутрибрюшных поражениях дифферециальный
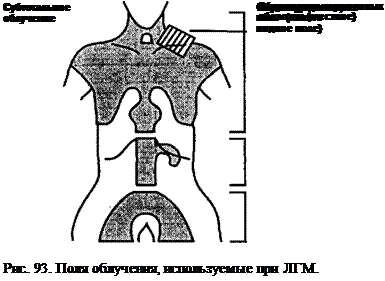
Тотальное облучение
Перевернутое Y-образное поле
диагноз проводят с туберкулезным мезаденитом, псевдотуберкулезом, неход-жкинскими лимфомами, а при гепатоспленомегалии — с болезнями накопления, портальной гипертензией, хроническим гепатитом, циррозом печени, опухолями. Лечение. При лечении больных ЛГМ применяют три варианта терапии: облучение, полихимиотерапию и комбинацию облучения и химиотерапии. При ЛГМ 1А/ ПА стадии рекомендуют субтотальное облучение лимфоузлов; IB/IIB стадии — комбинированную терапию; ША стадии — полихимиотерапию. У больных с минимальным поражением брюшной полости, выявленным на лапаротомии, и стадией ША можно использовать субтотальное или тотальное облучение лимфатических узлов; стадией IIIB/IV — полихимиотерапию. Комбинированная терапия показана при опухоли средостения большой массы. Лучевую терапию (рис. 93) при ЛГМ используют в трех вариантах [Ма-зай Дж., Робинсон У., 1997]: 1. Субтотальное облучение лимфоузлов или лимфоидной ткани состоит из мантиевидного и лопатовидного полей. Обычно его назначают при лечении больных со стадиями 1АД1А, ПВ. Доза 3500-4000 рад. 2. Тотальное облучение лимфоузлов и лимфоидной ткани состоит из мантиевидного и перевернутого У-образного полей, и его назначают для лечения стадий ПВ и ША. 3. Облучение пораженных областей включает только области с доказанным поражением, и обычно его применяют в комбинации с химиотерапией. Доза 2000-2500 рад. Таблица 169 Схема ОППА терапии для девочек с ЛГМ.
Химиотерапию проводят в специализированном онкологическом или гематологическом отделении комбинацией препаратов. В качестве примера приводим схему немецкого профессора Г. Шеллонга (1990) (табл.169, 170). Перерыв между циклами равен двум неделям. Обычный курс — 6 циклов в течение 6 мес. Условием начала следующего цикла является число лейкоцитов более 2000 в мкл и число гранулоцитов более 500 в мкл. Химиотерапию прерывают при наслоении тяжелой инфекции. Лучевую терапию обычно проводят также через 2 нед после окончания курса химиотерапии. Больные со стадиями ЛГМ ШВ, ША, IV, кроме циклов ОППА и ОЕПА, получают 2 цикла ЦООП-терапии (циклофосфан, винкристин-онковин, прокарбазин и преднизолон). С каждым введением циклофосфамида назначают уропротектор — урометоксан по 200 мг/м2 внутривенно струйно, сразу после введения циклофосфамида, а также через 4 и 8 часов. Используют и другие курсы комбинированной полихимиотерапии: МОПП (мустарген, винкристин-онкопин, прокарбазин-натулан, преднизолон), АБВД (адриамицин, блеомицин, винбластин, дакарбазин), ХВПП (хлорамбуцил, винбластин, прокарбазин, преднизолон) и другие. Осложнениями терапии у больных ЛГМ могут быть: гипотиреоидизм, стерильность и бесплодие, кардиотоксичность, асептический некроз головки бедра, вторичные инфекции и вторичные опухоли (нелимфобластная лейкемия, неходжкинские лимфомы, эпителиальные опухоли и саркомы), нейропатии, миелопатии. Прогноз. Прежде всего зависит от того, в какой стадии начато лечение. При локальных формах ЛГМ (IA, НА) полное выздоровление возможно у 70-80% детей, хотя полную ремиссию достигают у 90%. О выздоровлении можно говорить лишь через 10 лет после окончания успешного курса первичного лечения. Большинство рецидивов возникает в первые 3-4 года после окончания терапии. Объем контрольного обследования больных по Дж. Мазай и У. Робин-i сон (1997) следующий. Анамнез и физикальное обследование — каждый визит. Полный анализ крови — каждый визит. Биохимический анализ крови — каждые 6 мес в течение 4 лет (тесты, оценивающие функцию печени, уровни альбуминов и АДГ). '. Рентгенография грудной клетки — 1 раз в год в течение 10 лет. Компьютерная томография пораженных областей — 1 раз в год в течение 4 лет. Функциональное исследование щитовидной железы — 1 раз в год.
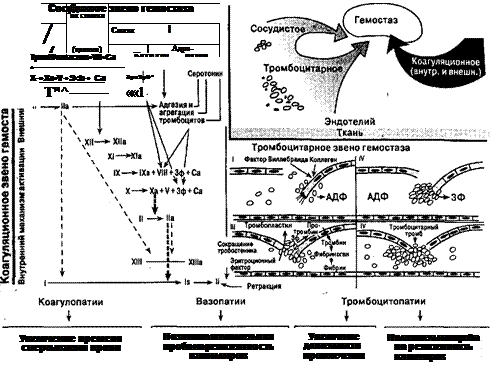
ГЕМОРРАГИЧЕСКИЕ ДИАТЕЗЫ И ГЕМОРРАГИЧЕСКИЕ ЗАБОЛЕВАНИЯ. Гемостаз — функциональная система организма, обеспечивающая, с одной стороны, остановку и предупреждение кровотечений при нарушении целостности сосудистой стенки, а с другой — сохранение жидкого состояния циркулирующей и депонированной крови. Кроме того, среди задач системы гемостаза выделяют регуляцию транскапиллярного обмена, резистентности и проницаемости сосудистой стенки, а также влияние на состояние репара-тивных процессов. В выполнении двух Противоположных задач (тромбиро-вание в месте повреждения сосуда и предупреждение тромбообразования в системном кровотоке) участвуют 3 звена — сосудистое, тромбоцитарное и плазменное, каждое из которых имеет элементы системы, способствующие образованию сгустка (коагулянты, точнее — прокоагулянты) и препятствующие этому процессу (антисвертывающие и фибринолитические факторы). При этом поддержание нормальной целостности и проницаемости сосудистой стенки (как в месте повреждения сосуда, так и в предупреждении тромбообразования) обеспечивается тесной взаимосвязью про- и антикоагулянтной, про- и антифибринолитической активности сосудистого, тромбоцитарного и плазменного звеньев. На рис. 94, 95 представлены основные узлы взаимодействия свертывающей, антисвертывающей и фибринолитической (плазмино-вой) систем в плазменном звене гемостаза. Для лучшего усвоения изложенного в данном разделе материала полезно по учебникам физиологии и патофизиологии освежить свои знания о гемостатических механизмах. Современный этап развития гемостазиологии характеризуется: 1. Интенсивным изучением интимных механизмов взаимодействия коа-гуляционного, тромбоцитарного и сосудистого компонентов системы гемостаза. Прежде всего имеется в виду тот факт, что тромбоциты несут клеточный пул про- и антикоагулянтов, роль которых не вполне ясна. Внутренняя активация Поверхность контакта г ATIII i XI ---- ► Xla
ATIII
Внешняя активация
Повреждение тканей
IX ---- *■ IXa + VIII + TO + Ca++ Ca++ + TO + Vlla VII
ATIII Протеин С ATIII
Протеин С
Протромбин ___ * > Тромбин ATIII
Фибриноген Продукты Фибрин------- ^ деградации >, фибрина , ATIII Плазминоген -*■ Плазмин
Активаторы плазминогена
Рис. 94. Схема гемостаза (И. Витт). а2-антиплазмин
Кровоизлияния по гема-томномутипу (гемартрозы и посттравматические кровотечения) Кровоизлияния по пурпурному типу (симметричные отграниченные, округлые высыпания при анафилактоидной пурпуре) Кровоизлияния по капиллярному или микроцирку ляторному типу (петехиально-пятнистые кровоизлияния в кожу и слизистые оболочки)
Рис. 95. Патология гемостаза. 2. Центральной фигурой гемостаза, как ауторегулируемого процесса, является тромбоцит, осуществляющий взаимосвязь эндотелия сосудов с плазменными белками, клетками крови и выполняющий ряд негемо-статических функций: регуляцию тканевого роста, ангиогенеза, пролиферации астроглии и других. Имеется также точка зрения о ведущей роли эндотелиоцитов в поддержании гомеостатического баланса гемостаза. 3. Рецепторный аппарат тромбоцитов, включающий селектиновое, интегри-новое и иммуноглобулиновое суперсемейства, подключает тромбоцит и его компоненты в общую медиаторную систему гемостаза, иммунитета и воспаления, что требует учета при анализе параметров гемостаза на фоне инфекционно-септического процесса, оперативных воздействий и иммунологического конфликта. Сформулирована новая концепция интимных тромбоцитарно-коагуляци-онных связей, которая предполагает специфическое активное вовлечение тромбоцитов в каталитические и защитные функции на всех этапах внутреннего пути активации коагуляции. При этом рассматривают два аспекта: влияние тромбоцитов на коагуляцию и влияние коагуляции на функции тромбоцитов. Ряд белков-коагулянтов связываются специфическими рецепторами мембраны тромбоцитов (факторы X, XI, XIa, XIII, фибриноген, тромбин). Четыре коагуляционных кофактора/субстрата содержатся в а-гранулах (фибриноген, фактор Виллебранда — (ФВ), V фактор и высокомолекулярный ки-ниноген) и четыре ингибитора плазменных протеиназ (а,-антитрипсин, а2-макроглобулин, С1ИН и ос2-антиплазмин) находятся в тромбоцитах, при этом плазменный ФВ и пластиночный ФВ связываются на тромбоцитах в различных участках. Клиническим следствием из изложенного является то, что нетяжелое повреждение или недостаточность одного из гемостатических механизмов (наследственная или приобретенная) может быть частично компенсирована усиленной функциональной активностью другого, а потому клинически в течение длительного периода жизни не проявляться повышенной кровоточивостью. Лишь когда появляется дополнительный возмущающий фактор (инфекционное заболевание и назначенные в связи с ним медикаменты, гиповитаминозы и другие пищевые дефициты, декомпенсированный дисбак-териоз кишечника, обменные нарушения, обострение различных хронических заболеваний, неблагоприятные экологические влияния и др.), может появиться геморрагический синдром. Отдельные эпизоды повышенной кровоточивости могут быть отделены друг от друга у человека с наследственным дефектом гемостаза многими месяцами и даже годами, а затем вдруг появляться достаточно часто. Особенно типично это для наследственных дефектов свойств тромбоцитов — тромбоцитопатий и болезни Виллебранда. Отсюда и название — геморрагические диатезы. С другой стороны, такие типичные дефекты коагуляционного звена гемостаза, как гемофилии, при тяжелом дефиците факторов VIII и IX (менее 1% нормы) являются уже геморрагическими заболеваниями. Другой вывод, вытекающий из изложенного — крайняя важность для диагностики наследственных дефектов гемостаза тщательного сбора анамнеза со скрупулезным анализом минимальных признаков повышенной кровоточивости (периодические кожный геморрагический синдром, носовые кровотечения или кровотечения после травм, порезов, длительность менструальных кровотечений у женщин и др.) не только у самого больного в прошлом, но и у всех известных родителям родственников. Анамнестически необходимо выяснить и тип кровоточивости у больного и его родственников. Для нарушения тромбоцитарного звена гемостаза характерен микроцирку-ляторный, петехиально-пятнистый (синячковый) тип повышенной кровоточивости — спонтанно, преимущественно по ночам возникающие несимметричные кровоизлияния в кожу и слизистые оболочки, периодические носовые кровотечения и микрогематурия; удлиненные менструальные кровотечения у женщин, мелена, длительные кровотечения после малых хирургических операций (удаления зубов, адено-тонзиллэктомии и др.). Для расстройств коагуляционного звена гемостаза типичен гематомный тип повышенной кровоточивости (болезненные напряженные кровоизлияния В мягкие ткани, суставы обычно после травм, инъекций, длительные кровотечения из ран). Васкулитно-пурпурны й тип наблюдается при инфекционных и аллергических васкулитах (чаще симметричные, нецветущие эритематозные или геморрагические сыпи, нередко сочетающиеся с кишечным кровотечением, часто трансформирующиеся в ДВС-синдром), а ангиоматозный — при телеангио-эктазиях, ангиомах, артериовенозных шунтах (упорные, строго локализованные и обусловленные локальной сосудистой патологией кровотечения). Для некоторых геморрагических диатезов и заболеваний типична смешанная си-нячково-гематомная кровоточивость (болезнь Виллебранда, дефицит факторов протромбинового комплекса и XIII фактора, ДВС-синдром, передозировка антикоагулянтов и тромболитиков, появление в крови иммунных ингибиторов XIII и IV факторов). Важно отметить, что тромбофилия — склонность к тромбозам, типична не Только для первичных (наследственных) и приобретенных (вторичных) дефицитов антикоагулянтов (антитромбина III, протеина С), плазминогена и его активаторов, но и для некоторых наследственных коагулопатий — дис-фибриногенемий, дефицита XII фактора, прекалликреина, Cj-эстеразного ингибитора, рикошетных тромбозов вследствие резкой отмены антикоагулянтов (например, гепарина) и тромболитической терапии (стрептокиназа, уреа-за и др.). Сравнительно недавно выяснено, что к тромбофилии предрасполагает на-' следственный дефект V фактора свертывания крови (Лейденовская мутация), мутационное повреждение гена метилентетрагидрофолатредуктазы, приводящее к повышению уровня гомоцистеина, дефект генов белков С, S, тромбомо-дулина, протромбина, II и VIII факторов свертывания крови и т. д. Зачастую эти генетические дефекты не проявляются, однако при определенных обстоятельствах они определяют развитие тромбозов. По данным лаборатории Е. И. Шварца, в Санкт-Петербурге лица с наследственным дефектом V фактора свертывания крови составляют 2% популяции; они имеют вероятность тромботического эпизода в 5-10 раз выше, чем в контроле, так как аномальный V фактор резистентен к протеину С (см. рис. 94). В то же время, если женщина с этим генотипом принимает гормональные контрацептивы, риск развития тромбозов увеличивается в 30 раз. Около 20% тромбозов легочной артерии — это дефект V фактора свертывания крови. Учитывая высокую частоту этого мутационного повреждения (от 2 до 6%) в разных европейских популяциях, предполагают ввести ДНК-диагностику данного дефекта у всех больных, подлежащих оперативному вмешательству. Одновременная склонность и к тромбообразованию, и к кровотечениям из-за гипокоагуляции — типичная черта синдрома диссеминированного внутри-сосудистого свертывания крови (ДВС-синдрома). Все первичные геморрагические диатезы и заболевания делят на 3 группы: коагулопатии, тромбоцитопатии и тромбоцитопении, вазопатии. По частоте этих заболеваний до недавнего времени считали, что доминируют нарушения тромбоцитарного звена гемостаза — приблизительно 2/3 всех больных с первичными дефектами гемостаза. В настоящее время, по мере совершенствования диагностики, увеличивается частота выявления болезни Виллебранда, и есть мнение, что в Европе она распространена с частотой 1% [Папаян Л. П., 1996]. В учебнике нет возможности даже кратко описать все наследственные дефекты гемостаза, ибо в настоящее время их насчитывают более 200. Вероятно, около 50 млн человек на Земле имеют первичные дефекты системы гемостаза.
Наследственные коагулопатии. Среди всех больных с наследственными коагулопатиями у 94-96% диагностируют гемофилию А и В, болезнь Виллебранда. Остальные формы (гемофилия С — дефицит XI фактора, болезнь Хагемана — дефицит XII фактора, парагемофилия — дефицит V фактора, диспротромбинемии — дефицит II, VII, X факторов, а также афибриногенемия, дисфибриногенемия, афибри-наземия — дефицит XIII фактора) встречаются редко и в учебнике описаны лишь как имеющие значение для дифференциальной диагностики. Комитет экспертов ВОЗ считает, что в Европейских странах приблизительно 1 из 10 ООО новорожденных мальчиков имеет тяжелую гемофилию (в этой группе гемофилия А встречается в 5 раз чаще, чем гемофилия В) и примерно 1-2 на 10 000 мужчин — средней тяжести и легкую гемофилию (частота гемо-г филии А и В одинакова). Суммарная частота наследственных дефектов I, II, V, VII, X, XI и XIII факторов равна 5-6 на 1 млн человек. В России примерно 15 000 больных гемофилией, из них 80-85% страдают гемофилией А и 15-20% — гемофилией В.
Гемофилия. Гемофилия — наследственная болезнь, передаваемая по рецессивному, сцепленному с Х-хромосомой типу, характеризующаяся резко замедленной свертываемостью крови и повышенной кровоточивостью из-за недостаточной коагуляционной активности VIII или IX плазменных факторов свертывания крови. Болеют лица мужского пола. Гемофилию В ранее называли болезнью Кристмасс. Патогенез Концентрация VIII и IX факторов в плазме крови невелика (соответственно 1-2 мг и 0,3-0,4 мг на 100 мл или одна молекула VIII фактора на 1 млн молекул альбумина), но при отсутствии одного из них свертывание крови в первую его фазу по внутреннему пути активации (рис. 94) очень резко замедляется или совсем не происходит. Дата добавления: 2014-10-03 | Просмотры: 1372 | Нарушение авторских прав |

 ATIII
ATIII