|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
Генез наиболее важных синдромов при ХПН. 7 страница
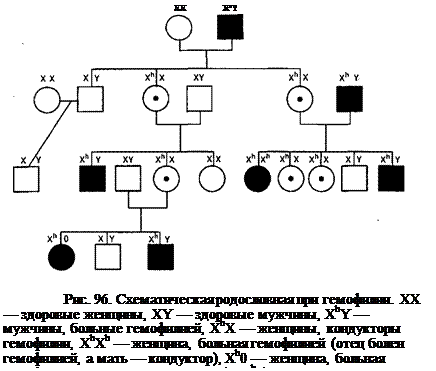
После выделения в 1980 г. Э. Тадденхемом чистого VIII фактора человека выяснено, что это — крупномолекулярный белок с массой 1 120 ООО дальтон, состоящий из ряда субъединиц с массой от 195 ООО до 240 ООО дальтон. Одна из этих субъединиц обладает коагуляционной активностью (VIII:K); другая — активностью фактора, отсутствующего при болезни Виллебранда и определяющего способность тромбоцитов агрегировать с ристоцетином, а также необходимого для их адгезии к поврежденной сосудистой стенке (VIII:OB); и по крайней мере две субъединицы, от которых зависит антигенная активность обеих субъединиц (VIII:Kar и VIILOBar). Синтез субъединиц VIII фактора происходит в разных местах: VIII:OB в эндотелии сосудов, a VIII:K, вероятно, в лимфоцитах. Где и как происходит сборка разных субъединиц в единую молекулу — неизвестно, хотя установлено, что в единой молекуле VIII фактора полимеризованных субъединиц VIII:OB несколько. У больных гемофилией А активность VIII:K резко снижена, тогда как концентрация VIII:Kar (при использовании кроличьих и козлиных антител к VIII:K человека, VIII:OB) нормальная. Значит, при гемофилии синтезируется аномальный VIII или IX факторы, не выполняющие коагуляционных функций. Ген, кодирующий синтез обоих белков, имеющих отношение к коагуляции (VIII:K, VIILKar), локализован на Х-хромосоме (Xq28), тогда как ген, определяющий синтез VIII:OB — на 12-й хромосоме. Ген VIILK выделен в 1984 г., состоит из 186 тыс. оснований. Создан ДНК-зонд, имитирующий характерную последовательность оснований этого гена, позволяющий с помощью блот-гибритизации выявлять наличие гемофильного гена в клетках человека и фибробластах околоплодных вод. Это дает возможность обнаруживать женщин-носителей гена, а также диагностировать гемофилию А у зародыша человека после 8 нед жизни. Р. М. Лон и Г. А. Вихар сообщили, что проанализировано 200 генов гемофилии и найдено среди них 7 разных мутаций, специфичных для каждой семьи. Этим методом было подтверждено, что примерно у 25% больных гемофилия — следствие спонтанной мутации. Рассчитана частота спонтанного мутирования: для гемофилии А она равна 1,3 х 10~5, а гемофилии В — 6 х 10~7. Наследование. Схематическая родословная больного гемофилией приведена на рис. 96. Все дочери больного гемофилией являются носителями гена гемофилии (кондукторами гемофилии), которые с вероятностью 1: 4 могут родить сына, больного гемофилией (гемофилия будет у 50% сыновей). Сыновья больного гемофилией здоровы и не могут передавать болезнь детям. Среди дочерей женщины-кондуктора гемофилии 50% также будут гетерозиготными носителями гемофильного гена. У женщин-носителей гемофильного гена обычно уровень VIII или IX факторов умеренно снижен до 30-50%, но кровоточивости нет, хотя она и может возникать после больших хирургических операций. Нечасто, но бывают женщины, симптоматические носители гемофильного гена, имеющие очень легкую склонность к кровоточивости.
Женщины, больные гемофилией, очень редки, и при выявлении низкого уровня VIII фактора в плазме у женщины с повышенной кровоточивостью надо прежде всего исключить болезнь Виллебранда, помня, что зачастую необходимы повторные обследования. Истинная гемофилия у женщин может быть если: 1) отец болен гемофилией, а мать — носитель гемофильного гена; 2) женщина-носитель гемофильного гена имеет синдром Тернера (ХО) или спонтанную мутацию, произошедшую в Х-хромосоме отца; 3) женщина-носитель гемофильного гена имеет синдром тестикулярной феминизации. Ингибиторная гемофилия. У 10% больных гемофилией при повторных переливаниях концентратов антигемофильных факторов, плазмы, крови возникают антитела к VIII или IX факторам, что существенно утяжеляет течение болезни, затрудняет лечение таких больных (ингибиторная гемофилия). Причины возникновения этого иммунопатологического процесса лишь у части больных (а также того, что у части больных гемофилией после повторных гемартрозов возникает артрит по типу ревматоидного) не ясны. Ингибиторная гемофилия может развиваться и у больных с различными иммунологически Клиническая картина. Характеризуется: 1) длительными кровотечениями после нарушения целостности кожных покровов и слизистых оболочек; 2) склонностью к очаговым массивным кровоизлияниям (гематомам) в подкожную клетчатку, мышцы, суставы, внутренние органы после минимальных травм, ударов и даже спонтанным кровоизлияниям (табл. 171). Отмечают определенную взаимосвязь между уровнем VIII фактора в крови и степенью тяжести кровоточивости (табл.172). Проявляется гемофилия чаще во второй половине 1-го — начале 2-го года жизни, но иногда позже или уже в неонатальном периоде. Кровотечения у больных гемофилией могут возникнуть не сразу после травм (например, экстракции зуба), а спустя У2-1 ч. Кровотечения длительные, обычно не останавливаются при местной гемостатической терапии. Даже такие процедуры, как взятие крови для анализа, подкожные и внутримышечные инъекции могут вызвать у больного кровотечение, продолжающееся часы и даже сутки. После внутримышечных инъекций типично возникновение очень обширных гематом, которые могут вызвать сдавление нервов, обусловливающее параличи и парезы. Кровоизлияния в суставы — самое характерное проявление гемофилии и наиболее частая причина инвалидизации больных. По частоте локализации Кровоизлияние в сустав обычно начинается спустя некоторое время после травмы (1 час и более): возникают острые боли, пораженный сустав увеличивается в объеме, кожа над ним горяча на ощупь, несколько блестящая, положение конечности флексорное. При вливании антигемофильной плазмы боль уже через несколько часов уменьшается. Недопустимо для уменьшения боли назначать нестероидные противовоспалительные средства, так как они могут усиливать повышенную кровоточивость за счет угнетения функции тромбоцитов. Однократное кровоизлияние в сустав предрасполагает к повторным именно в этом суставе, воспалительному процессу в нем, деструктивным и дистрофическим изменениям в окружающих хрящах и костях. В патогенезе хронического синовита, изменениях хрящей и костей, возникающих при повторных кровоизлияниях, важное значение имеют иммунопатологические процессы, которые у детей старше 12-13 лет и взрослых, больных гемофилией, уже могут привести к артриту, протекающему по типу ревматоидного, в мелких суставах кисти, стопы и других, где кровоизлияния не было. Частые гемартрозы предрасполагают также и к уролитиазу с кальциевыми камнями. После 15 лет тяжесть кровоточивости при гемофилии обычно ослабевает. Важно помнить, что нередко больным и их родителям необходима помощь психотерапевта, ибо тяжелое течение болезни — очень серьезная психологическая нагрузка для семьи. Диагноз и дифференциальный диагноз. Основан на анализе данных родословной (мужчины по материнской линии с кровоточивостью) и анамнеза, выявлении увеличения длительности свертывания венозной крови по Ли—Уайту (норма 8 мин, а при гемофилии — резко удлинено), увеличение времени рекальцификации плазмы и активированного частичного тромбопластинового времени (АЧТВ) и нарушения в первой фазе свертывания крови (при свертывании крови не весь протромбин уходит в сгусток, отсюда потребление протромбина низкое; норма 80-100%), низкого уровня VIII или IX фактора. Типичным изменением общих коагуляционных тестов у больных гемофилией является удлинение времени свертывания рекальцифицированной плазмы, замедление свертывания крови в активированном парциальном тромбо-пластиновом тесте (при добавлении имитаторов фосфолипидных мембран — коалина и кефалина) при нормальной длительности протромбинового теста Квика (время свертывания рекальцифицированной плазмы при добавлении тромбопластина человека или кролика) и тромбинового теста (время свертывания рекальцифицированной плазмы при добавлении тромбина). Точность ДНК-диагностики гемофилии — 78,2% (Баранов В. С). У больных с нарушениями тромбоцитарного звена гемостаза повышенная кровоточивость иная — кожный геморрагический синдром в виде петехий, небольших экхимозов (гематомы редко), гемартрозы для них нехарактерны, типичны рецидивирующие носовые кровотечения (при гемофилии это бывает редко), мелена, у девочек, достигших полового созревания, — маточные кровотечения. Обычно у этой группы больных кровотечения после небольших хирургических вмешательств (экстракция зуба, аденотомия и др.) возникают в момент операции, а не после нее, как при гемофилии. Родословные демонстрируют чаще доминантный тип наследования повышенной кровоточивости в семье. Эндотелиальные пробы (жгута, щипка и др.) положительные (чего не бывает при гемофилии), длительность кровотечения удлинена (что также нехарактерно для гемофилии). У больных с приобретенными вазопатиями отклонений в коагулогичес-ких тестах, как правило, нет (если отсутствует ДВС-синдром), среди родственников обычно не встречаются лица с повышенной кровоточивостью. При стертых формах гемофилии очень важно исключить болезнь Виллебранда (доминантный тип наследования кровоточивости, увеличение длительности кровотечения, резкое снижение адгезии тромбоцитов к стеклу и агрегации их с ристоцетином). Гемофилия С (болезнь Розенталя, дефицит XI фактора) характеризуется легким течением с признаками повышенной кровоточивости лишь после травм, хирургических вмешательств. Спонтанные кровотечения возникают редко (обычно носовые, менорра-гии). Гемартрозов, кровоизлияний в мозг, внутренние органы обычно не бывает. Тип наследования аутосомно-доминантный. Болеют как мужчины, так и женщины. Диагноз ставят на основании обнаружения типичных для гемофилии отклонений в первой фазе свертывания крови и дефицита XI фактора. Парагемофилия (болезнь Оврена, дефицит V фактора) наследуется по аутосомно-рецессивному типу и встречается лишь у гомозиготных носителей. Основное проявление — рецидивирующие носовые кровотечения, гематомы и кровотечения после небольших травм, у девочек — меноррагии (могут быть очень тяжелыми). Гемартрозы редки. Диагноз основан на выявлении дефекта свертывания крови как в первой, так и во второй фазе, низкого уровня V фактора. Лечение такое же, как при гемофилии А. Болезнь Стюарта — Проуэра (дефицит X фактора) наследуется по аутосомно-рецессивному типу и при тяжелом дефиците проявляется с рождения (кровотечения из пуповины и пупочной ранки), возникают кровотечения из мест травм, порезов. Гемартрозы редки. Диагноз основан на обнаружении дефекта свертывания крови в первой и второй фазах и низкого уровня X фактора. Лечение такое же, как и при гемофилии А. Дефициты протромбина и проконвертина (II, VII факторов) наследуются аутосомно-рецессивно и проявляются как средней тяжести гемофилия, но с выраженной спонтанной кровоточивостью слизистых оболочек (носовые, маточные, почечные, желудочно-кишечные кровотечения). Возможны кровоизлияния в суставы, внутренние органы, гематомы после травм. Диагноз основан на выявлении дефекта свертывания крови во второй фазе и низкого уровня II или VII фактора. Лечение — переливание плазмы, препарата PPSB (концентрата II, VII, IX, X факторов) 1 раз в 3-4 дня. Наследственный дефицит фактора Хагемана (XII фактор) наследуется по аутосомно-рецессивному или доминантному типу и геморрагическими расстройствами не проявляется (так же как и дефицит факторов Флетчера, Фитцжеральда). У некоторых больных имеется тенденция к тромбозам. Диагностируют при плановом анализе гемостаза перед операцией и при выявлении дефекта в первой фазе свертывания крови по удлинению времени активированного парциального тромбопластинового теста, дефициту XII фактора, прекалликреина, высокомолекулярного кининогена. Афибриногенемия, дисфибриногенемии наследуются по аутосомно-рецессивному типу (ген локализован на 4-й хромосоме). Клинические проявления кровоточивости варьируют: от отсутствия кровоточивости до появления уже в периоде новорожденное™ гематом, внутричерепных кровоизлияний, мелены, кровавой рвоты, кровотечения из ранки пуповины. В дальнейшем могут быть носовые кровотечения, кожный геморрагический синдром (пете-хии, экхимозы), желудочно-кишечные кровотечения, мышечные гематомы, удлиненные маточные кровотечения, а также большие потери крови после операций, то есть кровоточивость микроциркуляторного типа. Радикальный тест для диагностики — удлинение времени тромбинового теста и свертывания крови (иногда не сворачивается за много часов), сниженный уровень фибриногена (при дисфибриногенемиях, которых описано уже более 100 видов, концентрация фибриногена нормальная, но изменено его качество). Афибриназемия (отсутствие XIII фактора) наследуется как по аутосом-ному, так и сцепленному с Х-хромосомой рецессивному типу. Проявляется медленным заживлением пупочной ранки и кровотечением из нее. Уже на первом году жизни у половины больных развиваются кровоизлияния в мозг и его оболочки. Характерны также медленное заживление ран с образованием келоидных рубцов, желудочно-кишечные кровотечения, макрогематурия, кровоизлияния в брюшную полость. Кровоточивость смешанного микроцир-куляторно-гематомного типа. Все коагуляционные тесты нормальные (за исключением резкого удлинения времени растворения сгустков крови в 5 М растворе мочевины). Лечение — переливание свежезамороженной плазмы. Дефицит протеина С, витамина К-зависимого гликопротеина, активированная форма которого (активируется комплексом тромбин-тромбомодулин на сосудистой стенке) ингибирует активные формы V и VIII факторов и стимулирует фибринолиз, клинически выявляется уже в периоде новорожденное™ тяжелыми тромбозами в сочетании с кожным геморрагическим синдромом, то есть развитием диссеминированного внутрисосудистого свертывания крови, плохо управляемого гепарином. Показаны оральные антикоагулянты, анаболические стероиды (даназол), а в острый период — свежезамороженная плазма. Дефицит протеина S или антитромбина III, наследующийся также по аутосомному рецессивному типу, проявляется тромботической болезнью, диагностируют его при специальных исследованиях. Лечение. При гемофилии имеет следующие особенности: 1) внутримышечные инъекции запрещены (все препараты могут быть введены только внутривенно либо назначены внутрь); 2) кровотечение любой локализации и тяжести, припухлость и боль в суставе, подозрение на кровоизлияние во внутренние органы — показание к немедленному (даже ночью!) введению концентрированных антиге-мофильных препаратов; 3) аналогично следует поступить при травме с нарушением целостности кожных покровов; 4) больной должен раз в квартал посещать стоматолога, имеющего опыт лечения детей с гемофилией; 5) любые хирургические вмешательства возможны только после введения препаратов антигемофильного глобулина. Используют для вливаний лишь поверхностные вены. Применяют две программы лечения больных гемофилией: 1) систематическое трансфузионное лечение; 2) периодическое, симптоматическое трансфузионное лечение. Первая программа особенно показана детям лишь с тяжелой гемофилией: больным гемофилией А с 1 до 20 лет вводят криопреципитат в дозах 25-40 ЕД/кг 3 раза в неделю, а при гемофилии В концентрат IX фактора 25-40 ЕД/кг 2 раза в неделю. Такая терапия приводит через 1,5-2 года к 4-5-кратному увеличению уровня VIII и IX факторов и уменьшению тяжести течения болезни. Вторая программа предусматривает введение концентратов VIII и IX факторов в первые часы после кровоизлияния или обширной травмы. При гемофилии А внутривенно вводят криопреципитат в дозе 45-50 ЕД на 1 кг массы тела, а при гемофилии В — 35 ЕД/кг (концентрат IX фактора). Недостаточность для гемостатических целей использования свежезамороженной плазмы определяется тем, что обычно переливаемая доза ее 15 мл/кг (содержание VIII фактора — 0,6 ЕД/мл) позволяет увеличить уровень VIII фактора в крови больного гемофилией лишь на 15-20%, что недостаточно для остановки кровотечения. Криоконцентрат гораздо более эффективен, ибо переливание 1 ЕД его позволяет повысить уровень VIII фактора на 2%. Отсюда для остановки начинающегося кровоизлияния в сустав, при гематомах, небольших ранах, требующих наложения нескольких швов, достаточно перелить криопреципитат в дозе 25-30 ЕД/кг. При кровотечениях из языка, шеи, нижней части лица и дна полости рта уже необходимо начать вливания криопреципи-тата с 35-50 ЕД/кг. При внутричерепных кровоизлияниях, гематомах, сдавливающих нервы, необходим криопреципитат в дозе 50 ЕД/кг массы тела больного. Повторное введение в той же дозе надо сделать через 6-8 ч и далее два раза в день (4-5 дней) и ежедневно 1 раз в сутки (4-5 дней). Период полужизни VIII фактора при первом введении криопреципитата 4-8 ч, а при повторных — 12-36 ч. Повторные переливания любых препаратов крови очень опасны в плане инфицирования больного рядом вирусов (ВИЧ, гепатитов В, реже D, С, ци-томегалии, герпеса и др.). В США до 90% больных тяжелой гемофилией при обследовании в 1984 г. оказались инфицированными ВИЧ, 50-60% имели антитела к поверхностному антигену гепатита В. Больных гемофилией надо прививать против гепатита В. Безусловно, целесообразно и периодическое обследование всех больных гемофилией на эти инфекции. ВИЧ-инфекция у больных гемофилией детей сопровождается лимфоцитарным интерстициаль-ным пневмонитом, реже пневмоцистной пневмонией. Криопреципитат из пулированной крови доноров (сливают кровь приблизительно 2000 доноров) опасен при переливании больным А(П), В(Ш) и особенно AB(IV) групп крови в плане развития гемолиза из-за содержания в нем соответствующих изогемагглютининов. При необходимости экстракции зуба за 12-13 ч переливают концентрат VIII фактора в дозе 30-40 ЕД/кг, за 1 ч до операции — 40 ЕД/кг и через 4-10 4 после экстракции зуба 30-40 ЕД/кг и далее вливают ежедневно 3 дня, а затем через день до эпителизации лунки. Аналогично поступают и при других хирургических операциях, но первые две дозы криопреципитата (до и через 4-6 ч после операции) увеличивают до 50 ЕД/кг. Местная терапия: наложение тампонов с гемостатической губкой, тромбином, грудным молоком на место кровотечения. При необильных кровотечениях рекомендуют закапывать в нос десмопрессин; 4 раза в день и внутрь е-аминокапроновую кислоту по 50 мг/кг также 4 раза в день. При гемартрозах на 3-4 дня иммобилизуют сустав, накладывают эластический бинт (но не дольше). При очень выраженных, болезненных гемартрозах после переливания криопреципитата показана пункция сустава и удаление крови (хирургом!). Эффективность местного применения холода (льда) многими оспаривается. При гематуриях и повторных гемартрозах больным с III и большей степенью гемофильной артропатии показан 2-3-недельный курс преднизолоноте-рапии. При III-IV стадиях гемофильной артропатии обсуждают и вопрос о хирургическом лечении — синовэктомии. Рентгенотерапию рекомендуют для снижения частоты повторных кровоизлияний в сустав при III степени суставного процесса у детей старше 5 лет. Разовая доза — 0,5 Гр, суммарная — 4- 5 Гр. Рентгенотерапию проводят через день. В перспективе введение новых методов лечения — создание «миниорга-нов» — специфических линий гепатоцитов, которые после трансплантации под кожу больным будут синтезировать и поддерживать концентрацию в крови VIII фактора на уровне 15%. Возможен более многообещающий проект — имплантация нормальных генов факторов VIII или IX взамен дефектных. При ингибиторной гемофилии 3. С. Баркаган (1996) рекомендует три варианта терапии: 1) вливание в течение 3 дней рекомбинантного VIII фактора 100-300 ЕД/кг при угрожающих жизни кровотечениях: 2) вливание концентрата факторов протромбинового комплекса (PPSB); 3) вливание активированного VII фактора. Введение криопреципитата в больших дозах может подавлять синтез ингибитора. Для лечения инги-биторных форм в некоторых центрах успешно используют плазмаферез, а также вливания десмопрессина. Диспансерное наблюдение. Осуществляет совместно гематолог специализированного центра и участковый педиатр. Ребенка освобождают от прививок и занятий физкультурой в школе из-за опасности травм. Вместе с тем, физические нагрузки больному с гемофилией показаны, так как это увеличивает уровень VIII фактора. Питание больного ребенка не отличается от питания здоровых детей. Из лекарственных трав показаны отвары душицы и зайцегуба опьяняющего (лагохи-лус). При простудных заболеваниях не следует назначать ацетилсалициловую кислоту или индометацин (предпочтителен ацетоминофен). Противопоказаны банки, так как они могут спровоцировать возникновение легочных геморрагии. В воспитании ребенка важно акцентировать внимание на роли так называемых интеллектуальных профессий и постоянно стимулировать его интерес к чтению, нетравматическим развлечениям, вселять оптимизм. Полезны консультации психотерапевта. Прогноз При современных методах лечения значительно улучшился, и больные могут дожить до глубокой старости.
Болезнь Виллебранда. Болезнь Виллебранда (ангиогемофилия) — наследственное заболевание, передаваемое обычно по аутосомно-доминантному типу (рис. 97), характеризующееся повышенной кровоточивостью в сочетании с увеличением длительности кровотечения, количественным или качественным дефектом
2 3 4 DO 47 41 55
1 2 3 4 5 6 О 20 24 36 33 25
Рис. 97. Родословная больного 7 лет с болезнью Виллебранда. Восклицательный знак означает — лично обследован, заштрихованность — кровоточивость, зачерненность — лабораторное подтверждение диагноза. плазменного фактора Виллебранда (ФВ) и очень низкими величинами (или отсутствием) адгезии тромбоцитов к стеклу, агрегации тромбоцитов с ристо-цетином. Патогенез. Заболевание впервые описано Эриком фон Виллебрандом (1926) у девочки, проживающей на Аландских островах, у которой кровоточивость, протекающая по гематомному типу, сочеталась с низким уровнем в крови VIII фактора, резким удлинением времени кровотечения и доминантным типом наследования. Отсюда вытекало, что при описанной Виллебрандом болезни имеется коагуляционный дефект (низкий уровень VIII фактора) и сосудисто-тромбоцитарные нарушения (увеличение длительности кровотечения). Установлено, что синтез фактора Виллебранда регулируется аутосомным геном, расположенным на 12-й хромосоме, его нуклеотидная последовательность была определена в 1990 г. Фактор Виллебранда — белок-носитель для VIILK и необходим для адгезии (ретенции) тромбоцитов к микрофибриллам сосудистой стенки. В крови он находится в виде мультимеров с молекулярной массой от 500 тыс. до 2 млн дальтон. На стадии адгезии он связывает гликопротеин lb тромбоцитов с субэндотелиальными структурами поврежденной сосудистой стенки, а на стадии агрегации связывается с гликопротеином ПЬ/Ша поверхности тромбоцита. Основная масса ФВ синтезируется в специальных гранулах сосудистого эндотелия (тельца Паллади), выбрасываются в кровоток под влиянием тромбина, ионов кальция, 1-дезамино-8В-аргининвазопрессина (DDAVP). Именно в сосудистом эндотелии и находятся самые большие полимеры ФВ, которые и играют важнейшую роль в гемостазе. Описано 3 основных типа болезни Виллебранда: 1) частичный количественный дефицит (70-80% всех больных), 2) качественный дефицит ФВ с отсутствием больших мультимеров (10-12% всех больных, но при варианте ПА не образуются большие и промежуточные мультимеры; ПВ — имеется повышенная аффинность к гликопротеину lb тромбоцитов; IIM.N — снижена аффинность к VIILK или снижена функциональная активность тромбоцитов), 3) полный количественный дефицит VIII^B (3-5% больных). При всех вариантах в разной степени снижена агрегационная активность тромбоцитов с ристоцетином, хотя при ПВ типе она может быть иногда и нормальной. Сочетанность геморрагического дефекта определяет и вариабельность клинической картины с доминированием у одних больных коагуляционного, а у других — тромбоцитарного дефекта. Клиническая картина. При тяжелом течении болезни (уровень VIII фактора ниже 5% нормы) практически не отличается от проявлений гемофилии. При более высоком уровне VIII фактора на первый план выступает сосудисто-тромбоцитарпый тип кровоточивости: периодические обильные кожные геморрагии, носовые, маточные, желудочно-кишечные кровотечения (возможны и гематомы, в частности, на месте внутримышечных инъекций, гемартрозы). Кровотечения после небольших хирургических операций (экстракций зуба и др.) могут начинаться в момент вмешательства и продолжаться несколько суток, приводя к постгеморрагической анемии. После периода полового созревания тяжесть болезни уменьшается. Диагноз. Распознавание болезни возможно лишь при одновременном и динамическом изучении уровня VIII фактора и свойств тромбоцитов, потому что даже самые типичные для этого заболевания дефекты периодически могут исчезать. К характерным дефектам относят: 1) очень резкое увеличение длительности кровотечения; 2) низкая адгезия тромбоцитов к стеклонитям или стеклянным бусинам; 3) малое количество VIII фактора крови; 4) низкая агрегация тромбоцитов с ристоцетином. Последний дефект наиболее стойкий. Количество тромбоцитов всегда в норме. Эндотелиальные пробы могут быть как положительными, так и отрицательными. Очень важно при подозрении на болезнь Виллебранда обследовать больного в динамике, ибо даже при I типе болезни, наиболее изученном и распространенном, агрегация тромбоцитов с ристоцетином и уровень VIII фактора могут быть периодически нормальными. В специализированных центрах для диагностики используют определение в плазме крови VIII:K, VIir.OB, VIII:OBar, VIII:ФB-пoлимepa, агрегацию тромбоцитов с очень низкими дозами ристомицина (0,2-0,6 мг/мл), на которые отвечают тромбоциты здоровых лиц, а также больных гемофилией А, но не отвечают при I, ПА, III типе болезни Виллебранда. Зачастую диагноз возможен лишь при повторных обследованиях. Метод ДНК-диагностики болезни Виллебранда наиболее информативен и по В. С. Баранову его диагностическая ценность приближается к 100%. Синдром болезни Виллебранда может быть при диффузных болезнях соединительной ткани и другой иммунопатологии. Лечение. При легких и среднетяжелых формах I типа болезни, когда уровень VIII:K выше 5%, целесообразно назначение аналога естественного антидиуретического гормона — DDAVP (десмопрессина ацетат-1-деамино, 8-Б-аргинин-вазопрессин). DDAVP вводят внутривенно в дозе 0,3 мкг/кг массы тела больного в 30-50 мл изотонического раствора хлорида натрия в течение 10-20 мин. Синтез VIIP.OB и VIII:K, а также активатора плазминогена начинают повышаться уже через 30 мин, достигая максимума через 1У2-2 ч (у здоровых волонтеров 300-400%). Период полужизии. препарата в крови колеблется от 7 до 75 мин, но период подъема УШ:ФВ и VIII:K около 12 ч (после переливания криоконцентрата или свежезамороженной плазмы период полужизни УШ:ФВ равен 18 ч, a VIII:K — 29-48 ч). Отсюда ясно, что повторные введения необходимы через 12 ч, хотя в дальнейшем возможны и через 24 ч. 1 мкг DDAVP имеет активность 4 ИЕ антидиуретического гормона, а потому при повторных введениях возможны водная интоксикация и гипонатриемия. Побочные эффекты в виде транзиторной головной боли и болей в животе, покраснения лица, гипертензии обычно тяжелы и кратковременные При ПВ форме болезни Виллебранда вливание DDAVP может вызвать тромбоцитопению. При всех формах болезни Виллебранда (кроме тромбоцитарного) высокоэффективны как гемостатические средства вливания свежезамороженной плазмы (15 мл/кг) или криопреципитата, содержащих и VIII:OB. Длительность увеличения активности VIII:K фактора после вливания — 2-3 сут, показано при болезни Виллебранда периодическое назначение дицинона, пан-тотената кальция, фитотерапия. Прогноз Прогноз для жизни благоприятный.
Тромбоцитопеническая пурпура. Выделяют первичные и вторичные тромбоцитопенические пурпуры. К первичным относят: — идиопатическую тромбоцитопеническую пурпуру (болезнь Верльгофа); — наследственные; — изоиммунные (врожденная — при несовместимости плода и матери по тромбоцитарным антигенам, посттрансфузионная — после переливаний крови и тромбоцитной массы); Дата добавления: 2014-10-03 | Просмотры: 1256 | Нарушение авторских прав |