|
АкушерствоАнатомияАнестезиологияВакцинопрофилактикаВалеологияВетеринарияГигиенаЗаболеванияИммунологияКардиологияНеврологияНефрологияОнкологияОториноларингологияОфтальмологияПаразитологияПедиатрияПервая помощьПсихиатрияПульмонологияРеанимацияРевматологияСтоматологияТерапияТоксикологияТравматологияУрологияФармакологияФармацевтикаФизиотерапияФтизиатрияХирургияЭндокринологияЭпидемиология |
ГЛАВА 357. МЫШЕЧНАЯ ДИСТРОФИЯ И ДРУГИЕ ХРОНИЧЕСКИЕ МИОПАТИИ
Дж. Р. Менделл, Р. К. Григгс (J. R. Mendell, R. С. Criggs)
Большинство миопатий (см. табл. 354-2). включая наследственные, воспалительные, эндокринные, метаболические и токсические, могут проявляться хронической слабостью мышц. Принципы дифференциальной диагностики этих болезней суммированы в гл. 354.
Наследственные миопатии
Мышечные дистрофии
Термин «мышечная дистрофия» характеризует группу подчас совершенно разных болезней, сходство между которыми состоит в названии и в том, что они являются наследуемыми. Каждый тип мышечной дистрофии имеет свои особые фенотипические и генетические характеристики (табл. 357-1). Мышечная дистрофия Дюшенна. Это заболевание впервые было описано Edward Meryon (1852 г.), однако оно носит имя французского невролога Дюшенна. Дистрофия Дюшенна — это наследственная болезнь, наследование рецессивное, сцепленное с Х-хромосомой, чем объясняется преимущественное поражение лиц мужского пола. Частота заболевания колеблется от 13 до 33 на 100000 живорожденных младенцев мужского пола. У 1/3 пациентов в семейном анамнезе этого заболевания нет, что свидетельствует о возникновении многих случаев за счет новых мутаций. Тщательное обследование немногочисленных женщин с дистрофией Дюшенна (ДД) позволило предположить, что ген ДД локализуется в хромосоме. Так. у всех больных обнаруживают транслокацию и делецию на коротком плече Х-хромосомы в области Хр21. Доказательство близкого расположения локуса Дюшенна к области Хр21 было получено при изучении сцепления генов с использованием «ограничивающих» эндонуклеаз. Дальнейшее совершенствование методик генного анализа позволит идентифицировать плод с риском по данному заболеванию и создать методики прямого и непосредственного выявления носителей указанного гена. У женщин-носителей, гетерозиготных по данному гену, часто проявляются клинические черты заболевания,
Таблица 357-1. Прогрессирующие мышечные дистрофии
но современные методы его диагностики, такие, как определение сывороточной активности КК. пируваткиназы, лактатдегидрогеназы и другие, позволяют выявлять лишь 50% носителей этого наследственного заболевания. Клинически болезнь проявляется обычно в возрасте 3—5 лет. При этом больные мальчики часто падают и не могут подолгу играть со своими сверстниками в подвижные игры. Движения при беге, прыжки и подскакивания у них. как правило, отличаются от нормы. Те или иные двигательные нарушения могут быть замечены и в более раннем возрасте, но если нет семейного анамнеза, то на это не обращают внимания. К 5-летнему возрасту мышечная слабость становится совершенно очевидной, ребенок теряет способность совершать активные движения, прыгать, бегать Чтобы встать с пола, он должен упереться в пол руками — симптом Юверса. У маленьких детей икроножные мышцы обычно бывают гипертрофированы; в более позднем возрасте такое увеличение икроножных мышц следует называть псевдогипертрофией, поскольку мышечная ткань в них замещается жировой и соединительной. К 7—8-летнему возрасту становятся заметными контрактуры ахилловых сухожилий и илиотибиальных соединительнотканных пучков, при этом ходьба на пальцах ног заставляет больных откидываться назад, возникает лордозная поза. Потеря мышечной силы прогрессирует, преобладают поражения проксимальных мышц конечностей и сгибателей шеи; поражение нижних конечностей бывает обычно более резко выражено, чем верхних. В возрасте 8-10 лет больной уже не может передвигаться без помощи «вожжей»; контрактуры суставов и ограничение сгибания бедра и колена, локтя и разгибания кисти усугубляются длительным положением больного в сидячем положении. К 12 годам большинство больных передвигаются лишь в инвалидном кресле. Постепенно контрактуры становятся фиксированными, развивается прогрессирующий сколиоз. Деформация грудной клетки, обусловленная сколиозом, вызывает нарушение дыхательной функции легких, которая уже и без того снижена из-за слабости дыхательных мышц. К возрасту 14—18 лет у больных может развиться серьезная, иногда фатальная легочная инфекция. Другими причинами смерти могут послужить аспирация пищи и острое расширение желудка. Смерть от сердечной недостаточности у таких больных наступает нечасто, несмотря на наличие миокардиопатии. Застойная сердечная недостаточность возникает лишь при каких-то дополнительных стрессовых обстоятельствах, например при присоединении пневмонии. Сердечные аритмии отмечаются редко. В типичных случаях на ЭКГ отмечается углубление зубца S (RS) в отведении V1, глубокие и узкие зубцы Q в латеральных прекордиальных отведениях и RCR1 или полифазный зубец R в отведении V1. Интеллектуальные расстройства типичны при синдроме Дюшенна. У 1/3 больных коэффициент интеллекта обычно ниже 75, а в среднем равен 85. Причем снижение интеллекта не обусловлено мышечной слабостью, так как вербальные функции нарушаются обычно до наступления резко выраженной мышечной слабости. Причина интеллектуальных расстройств неясна. В отличие от мышечного поражения интеллектуальные нарушения не прогрессируют. Лабораторные исследования заключаются в определении сывороточной активности КК, которая, как правило, превышает норму в 20-100 раз. Активность этого фермента в крови бывает повышенной уже при рождении, что позволяет установить диагноз в самом начале жизни больного. Сывороточная активность КК остается повышенной в течение почти всей жизни больного и снижается лишь на поздних стадиях заболевания в связи со слишком малой мышечной активностью и потерей мышечной массы. Миопатию можно выявить при ЭМГ. В биоптате пораженной мышцы присутствуют мышечные волокна различного размера наряду с небольшими группами некротизированных и регенерирующих волокон. Потеря мышечных волокон обычно восполняется жировой и соединительной тканями. Мышечная дистрофия Беккера. Не такая тяжелая форма мышечной дистрофии. наследуемой по рецессивному типу и сцепленной с Х-хромосомой, была описана Беккером и Кейнером (Becker. Keiner) в 1955 г. Болезнь нередко называют доброкачественной формой псевдогипертрофической мышечной дистрофии. Клинические ее проявления весьма сходны с таковыми миодистрофии Дюшенна, однако прогрессирует она более медленно. Частота этого заболевания составляет примерно 1/10 от таковой миодистрофии Дюшенна. До 5-летнего возраста заболевание обычно не распознается, больные сохраняют способность ходить после 15 лет, а изредка и после 30 лет. Обращает на себя внимание резко выраженная гипертрофия икроножных мышц. После 40-летнего возраста больной может погибнуть от тех же осложнений, что и при миодистрофии Дюшенна. Тот факт, что локусы генов этих двух дистрофий (Дюшенна и Беккера) расположены очень близко друг от друга на Х-хромосоме, заставляет предполагать, что указанные болезни составляют аллельную пару. Методы выявления носителей указанных генов идентичны при этих двух заболеваниях и имеют одни и те же недостатки. В отличие от больных с дистрофией Дюшенна больные с миодистрофией Беккера могут достигать детородного возраста. При этом следует помнить, что наследуют миодистрофию Беккера только дочери (но не сыновья) — все они являются носителями этого гена. Лабораторное подтверждение диагноза включает определение сывороточной активности КК — динамика ее аналогична таковой при дистрофии Дюшенна. Аналогичны и изменения на ЭМГ и в биоптате пораженной мышцы. Плече-лопаточно-лицевая мышечная дистрофия (ПЛЛД). Это медленно прогрессирующая относительно доброкачественная миодистрофия, наследуется по аутосомно-доминантному типу. Женщины и мужчины болеют в равной степени. Течение ее очень вариабельно. Болезнь может начаться в любом возрасте, обычно в 3—4-м десятилетии жизни. Однако у некоторых больных симптомы могут не проявляться вовсе. Как следует из названия, обычно поражаются мышцы лица, плечевого пояса и проксимальные мышцы верхней конечности. Отставание лопатки от спины и свисание соответствующего плеча говорят о слабости передней зубчатой, трапециевидной и ромбовидной мышц; позже в патологический процесс вовлекаются двуглавая и трехглавая мышцы; дельтовидная мышца часто остается интактной. При поражении мышц лица у больных скудная мимика, угрюмое выражение лица, они не могут свистеть. В связи со слабостью передней большеберцовой и малоберцовой мышц свисание стопы может возникнуть уже на ранних стадиях заболевания. Иногда слабость мышц конечностей может прогрессировать до потери больным способности передвигаться самостоятельно. Другие системы организма при ППЛД обычно не поражаются. Сердце и органы дыхания вовлекаются редко, и в таких случаях следует думать о присоединении какого-либо другого заболевания. Очень часто при взгляде на больного создается впечатление о наличии у него экзофтальма, однако функция щитовидной -железы при этом остается нормальной. Характерна мягкая и лабильная артериальная гипертензия. Интеллектуальные функции обычно не нарушаются, так что больной сохраняет нормальный образ жизни. Диагностические исследования в типичных случаях излишни, особенно когда имеется отягощенная наследственность по данному заболеванию. Активность сывороточного КК может быть нормальной или слегка повышенной. Изменения на ЭМГ и в мышечном биоптате часто носят смешанный характер, говорящий и в пользу миопатии, и в пользу невропатии, что может повести к ошибочному диагнозу. Специфического лечения нет: при «свисающей стопе» целесообразно применение ортопедических аппаратов на область голеностопного сустава. Мероприятия, направленные на стабилизацию лопатки, могут уменьшить ее «отставание», но практически не улучшают ее функцию. Конечностно-поясная мышечная дистрофия (КПМД). Уже из названия болезни видно, что это сочетанное расстройство функций нескольких групп мышц. Поскольку наследование носит аутосомно-рецессивный характер, заболевание встречается спорадически. Слабость проксимальных мышц может начаться либо в верхних конечностях, либо в нижних, но обычно при прогрессировании патологического процесса поражаются все четыре конечности. Мышечная слабость может проявиться уже в детстве (в возрасте до пяти лет) или же позже: например, в 3-м десятилетии жизни; иногда она сопровождается псевдогипертрофией икроножных и других мышц. После первых проявлений заболевания больной может сохранить способность ходить более 20 лет. У некоторых больных в патологический процесс вовлекается сердце, что может привести к развитию застойной сердечной недостаточности или аритмиям. В отдельных случаях заболевание само по себе может проявляться как кардиомиопатия. По истечении тридцати или более лет к основным признакам болезни присоединяется дыхательная недостаточность. Интеллектуальные функции не страдают. При дифференциальной диагностике необходимо исключить воспалительные и метаболические миопатии, а также фенотипически сходные спинальные мышечные атрофии. Активность сывороточной КК несколько повышена, но не в такой степени, как при миодистрофиях Дюшенна или Беккера. На ЭМГ выявляются признаки миопатии, а в мышечном биоптате — признаки активной миопатии. Миотоническая дистрофия. Это заболевание наследуется по аутосомно-доминантному типу и поражает мышечную и другие ткани. Частота его составляет 1:10 000, но цифра эта несколько занижена, так как часто болезнь остается недиагностированной. Миотоническая дистрофия часто сопровождается интеллектуальными нарушениями, гиперсомнией, поражениями сердца, катарактой, атрофией гонад, дыхательной недостаточностью и поражением желудочно-кишечного тракта. Вначале слабость появляется в мышцах век, височных и лицевых, а также в сгибателях шеи и в дистальных мышцах конечностей. Миотонию выявляют на основании хватательной пробы, пробы на разгибание кисти или пробы на мышцы тенара, или при перкуссии языка. Заболевание начинается обычно в возрасте 10—20 лет, но клинически оно может не проявиться вообще. Дети, рожденные от матерей с миотонической дистрофией, могут страдать тяжелой формой этого заболевания, называемой врожденной миотонической дистрофией, характеризующейся резкой слабостью лицевых и бульбарных мышц; может наблюдаться также неонатальная дыхательная недостаточность самолимитирующего характера. У больных детей часто снижен интеллект. Диагноз часто основывается на характерных клинических признаках: слабость лицевых мышц обусловливает феномен типичного узкого, как бы выгравированного лица; отмечается преждевременное фронтальное облысение. При выявлении слабости дистальных мышц и миотонии диагноз подтверждается. Необходимости в лабораторных исследованиях при этом нет; более того. они могут привести к ошибочным путям диагностического поиска. Сывороточная активность КК обычно нормальная или слегка повышена. При ЭМГ с дистальных мышц кистей выявляют миотонию и миопатию. В биоптате пораженной мышцы обнаруживают четко выраженную атрофию мышечных волокон I типа, а в тяжело пораженных мышцах находят мышечные волокна кольцевидной формы, саркоплазматические массы и большое число центрально расположенных ядер. При вовлечении в патологический процесс сердца обычно страдает его проводящая система; у большинства бальных диагностируют блокаду I степени, а при полной поперечной блокаде необходима имплантация искусственного водителя ритма. Поскольку нарушения внутрисердечной проводимости чреваты внезапной смертью, то больным с указанными расстройствами следует настоятельно рекомендовать тщательное мониторирование, хотя точных временных критериев для имплантации водителя ритма не установлено. Реже возникают тахиаритмии и застойная сердечная недостаточность. У больных с незначительной слабостью мышц конечностей иногда отмечают довольно резко выраженную слабость дыхательной мускулатуры. Нарушение вентиляционной функции легких в сочетании с повышенной чувствительностью к депрессантам (в частности, угнетающим дыхательный центр) может привести к внезапно развивающейся дыхательной недостаточности в случае применения больным даже небольших доз опиатов и других седативных средств, особенно в пред- или постоперационном периоде. Определенную опасность представляет возможность возникновения эпизодов апноэ во время сна как центрального, так и периферического происхождения (см. гл. 215). Хроническая гипоксия может вести к развитию легочного сердца, что очень часто служит причиной появления сердечной недостаточности у подобных больных. Больным с миотонией редко требуется лечение по поводу именно этого состояния. поскольку болезнь редко достигает такой выраженности. Препаратом выбора в подобных случаях является фенитоин, так как другие антимиотические агенты — хинин и новокаинамид — могут еще более ухудшить сердечную проводимость. Мутантным геном миотоническая дистрофия может передаваться на хромосому 19, которая связана с генами, способствующими секреции определенных субстанций: изоантигены системы Лютеран (система групп крови Лютеран). пептидаза D и третий компонент комплемента. Использование методики сцепления генов позволяет в отдельных семьях своевременно устанавливать диагноз или даже осуществлять антенатальную диагностику. И более того, еще до появления тех или иных симптомов заболевания с помощью клинических и ЭМГ-исследований у членов пораженной семьи можно выявить скрытую форму миотонии, а при обследовании со щелевой лампой — обнаружить характерную заднюю субкапсулярную катаракту. Врожденная миотония. Болезнь наследуется как по аутосомно-доминантному (болезнь Томсена). так и по аутосомно-рецессивному типу (см. гл. 17). У больных с аутосомно-рецессивной формой наблюдают нерезко выраженную слабость, чего не бывает у больных с доминантной формой врожденной миотонии. Для лечения таких больных наиболее эффективны хинин, новокаинамид, фенитоин или диакарб. Поражений сердца не зарегистрировано. Окулофарингеальная дистрофия. Термином «прогрессирующая наружная офтальмоплегия» характеризуют состояние, сопровождающееся медленно прогрессирующим птозом и ограничением движений глазного яблока без вовлечения в патологический процесс мышц зрачка и нарушений аккомодации. Больные обычно не предъявляют жалоб на диплопию в отличие от случаев с более острым началом развития слабости глазодвигательных мышц. Окулофарингеальная дистрофия —это аутосомно-доминантное заболевание, при котором офтальмоплегия проявляется лишь в 5—6-м десятилетиях жизни. Многие больные являются потомками французско-канадской или испанской генеалогической ветви. Слабость фарингеальных мышц приводит к крикофарингеальной ахалазии, прогрессирующим затруднениям при глотании и нередко асимптоматической аспирации пищи в дыхательные пути. Если больных не лечить, у них могут возникнуть серьезные расстройства питания; лечение — хирургическая коррекция крикофарингеальной ахалазии. Помимо этого, окулярные миопатии могут быть обусловлены эффектом митохондрий в мышцах (см. ниже). Врожденная мышечная дистрофия. Начальные симптомы этого редко встречающегося заболевания могут сопровождать несколько нозологических единиц. В типичных случаях гипотония проявляется еще в младенческом возрасте, характерны уменьшение мышц в объеме наряду с контрактурами суставов конечностей. Активность сывороточного КК повышена, а при биопсии пораженных мышц обнаруживается типичная мышечная дистрофия. Заболевание носит относительно непрогрессирующий характер, однако многие больные не могут передвигаться самостоятельно. Позднее присоединяется дыхательная недостаточность. С помощью КТ у некоторых больных выявляют гипомиелинизацию глубоко лежащих слоев белого вещества мозга, однако это не имеет каких-нибудь известных клинических проявлений. Дистальная мышечная дистрофия. У этой редкой формы заболевания по крайней мере три отдельных варианта. Чаще всего встречается аутосомно-рецессивный, или спорадический вариант, который проявляется слабостью дистальных мышц конечностей во 2—3-м десятилетиях жизни. Постепенно мышечная слабость распространяется на более проксимальные мышцы. Активность сывороточной КК резко повышена. К другим вариантам дистальной миопатии относится аутосомно-доминантный, скандинавский вариант (форма миопатии Виландера), который начинается с поражения кистей, и аутосомно-доминантный вариант с поздним началом (4—5-е десятилетия жизни), который начинается с поражения нижних конечностей и довольно часто сопровождается кардиомиопатией. Скапулоперонеальная дистрофия. Несколько форм нервно-мышечной дистрофии вызывают свисание стопы и отставание лопатки от тела. Аутосомно-доминантная форма этого заболевания проявляется в 3—4-м десятилетиях жизни, она очень вариабельна по своему течению; дыхательная недостаточность встречается довольно редко, а кардиомиопатия — часто. Рецессивная форма заболевания, связанная с Х-хромосомой (форма Ешегу — Dreifuss), начинается в раннем детстве и характеризуется резко выраженными контрактурами суставов и нарушениями внутрисердечной проводимости. Иногда плече-лопаточно-лицевая дистрофия (если не проявляется слабость лицевой мускулатуры), очень напоминает скапулоперонеальную дистрофию.
Врожденные миопатии
Эти редко встречающиеся заболевания отличаются от мышечных дистрофий наличием специфических гистобиохимических и структурных дефектов мышечной ткани. Заболевание характеризуется непрогрессирующим течением, что, однако, не является правилом. В типичных случаях у младенцев отмечают гипотонию и задержку двигательного развития. Часто наблюдают «грудь сапожника», кифосколиоз, неправильное положение головки тазовой кости и вогнутую стопу (pes cavus). Своевременная диагностика очень важна, так как долгосрочный прогноз и лечение отличаются от таковых при мышечных дистрофиях. Различают четыре главные формы врожденной миопатии: центрально-стволовую, немалиновую (стержневая) миопатию, миотубулярную (центронуклеарная) миопатию и врожденную диспропорцию волокнистого типа. Центрально-стволовая болезнь. Это первая из описанных врожденных миопатий, идентифицированная Shy и Magee в 1956 г. Заболевание наследуется по аутосомно-доминантному типу, однако бывают и спорадические случаи. В младенческом возрасте характерными являются гипотония и задержка двигательного развития, однако заболевание может привлечь к себе внимание и лишь во взрослом возрасте, когда появляется мышечная слабость или те или иные изменения скелета. Больные небольшого роста с хрупкой фигурой; аномалии скелета характеризуются врожденной дислокацией бедер, сколиозом, вогнутой стопой (pes cavus) и «грудью сапожника». Слабость мышц лица и конечностей (особенно конечностей) выражена не очень резко. При исследовании биоптата мышцы обнаруживают мышечные волокна с множественными или единичными прерывистыми (дискретными) зонами (центральные массы некротизированной ткани), лишенные окислительных ферментов. Другие лабораторные тесты менее информативны диагностически, так как активность сывороточной КК и данные ЭМГ могут быть нормальными. Больные с этой патологией предрасположены к развитию злокачественной гипертермии (см. гл. 8). Немалиновая миопатия. Немалиновая миопатия, называемая также врожденной непрогрессирующей нитеобразной миопатией, была описана Shy и сотр. в 1963 г. Наследуется обычно по аутосомно-доминантному типу, но наследование может быть тоже рецессивным или спорадическим. В младенческом возрасте часто появляется гипотония, смерть может наступить вследствие дыхательной недостаточности. Скелетные аномалии могут быть выражены очень резко. Это резко удлиненное лицо; высокое небо, плохо развитая мускулатура, кифосколиоз, «грудь сапожника», мышечная слабость может распространяться на лицо, мягкое небо, конечности. Прогноз заболевания весьма вариабелен: иногда болезнь не прогрессирует, а если прогрессирует, то вынуждает больных пользоваться сидячей каталкой или же приводит к дыхательной недостаточности. При гистологическом исследовании в мышцах обнаруживают пучки нитеподобных или палочкоподобных немалиновых телец, от чего и произошло название данного заболевания. «Палочки» являются дериватами Z-пучковой субстанции, их обычно находят в мышечных волокнах I типа. а в пораженных мышцах, как правило, преобладают мышечные волокна именно этого типа. Активность сывороточной КК может быть нормальной или слегка повышенной. При ЭМГ обычно выявляют миопатию. Миотубулярная миопатия. Заболевание это было описано Spiro. Shy и Gonatas в 1963 г. Гистологические изменения при миотубулярной миопатии напоминают эмбриональную стадию развития мышечных «трубочек» в процессе формирования мышечного волокна. Некоторые авторы предпочитают называть заболевание центронуклеарной миопатией, полагая, что мышечные волокна при этом не имеют эмбрионального характера. Заболевание обычно возникает спорадически, но наследование может быть аутосомно-доминантным, рецессивным или рецессивным сцепленным с Х-хромосомой. В младенческом возрасте отмечаются гипотония и мышечная слабость, что может служить причиной смерти. Проявляясь в более старшем возрасте, болезнь может напоминать немалиновую миопатию. У больных при этом бывают узкое, вытянутое лицо, вогнутая стопа, сколиоз. Общая мышечная масса обычно уменьшена. а степень слабости проксимальной и дистальной мускулатуры варьирует. От других врожденных миопатий эта отличается присутствием наружной офтальмоплегии. Течение болезни может быть как прогрессирующим, так и непрогрессирующим. Активность сывороточной КК нормальная или слегка повышена. ЭМГ патологически изменена: потенциалы моторных единиц уменьшены и избыточно «укомплектованы». В биоптате мышцы обнаруживают мышечные волокна с рядами центрально расположенных ядер, часто окруженных чистой перинуклеарной зоной. В большей степени поражаются мышечные волокна I типа, они могут подвергаться атрофии. Врожденная диспропорция в соотношении типов мышечных волокон. К клиническим проявлениям этого заболевания относят гипотонию, мышечную слабость, замедленное физическое развитие, а также аномалии скелета, как и при других врожденных миопатиях. Диагноз основан на результатах мышечной биопсии: в биоптате отмечают увеличение числа небольших мышечных волокон I типа и нормальные или гипертрофированные мышечные волокна II типа. Патогенез заболевания малопонятный. Прогноз обычно благоприятный, у многих бальных с возрастом состояние улучшается, хотя двигательные нарушения той или иной степени все же остаются. У отдельных больных мышечная слабость прогрессирует.
Болезни, обусловленные нарушением энергетического метаболизма в мышцах
В скелетной мускулатуре обычно утилизируются два главных источника энергии — жирные кислоты и глюкоза. Следовательно, нарушение утилизации глюкозы или жиров может сопровождаться четкими клиническими проявлениями со стороны мышечной системы. Наиболее тяжелым проявлением данной патологии является острый мышечный болевой синдром, который может привести к тяжелому рабдомиолизу и миоглобинурии. Следует упомянуть также прогрессирующую мышечную слабость, симулирующую мышечную дистрофию. Объяснений существования этих двух различных клинических синдромов нет. Гликогеноз (болезнь накопления гликогена) и гликолитические дефекты. Существует четыре вида нарушений обмена гликогена (типы II, III, IV и V) и четыре вида расстройства гликолиза (типы VII, IX, Х и XI), которые проявляются существенными нарушениями со стороны скелетной мускулатуры (см. также гл. 313). Недостаточность кислой мальтазы (тип II гликогеноза). Кислая мальтаза — это лизосомный энзим из группы кислых гидролаз, обладающий a-1,4 и a-1,6 глюкозидазной активностью: расщепляет гликоген до глюкозы. В то же время роль этого фермента в обмене углеводов определена недостаточно четко. Существует три клинические формы недостаточности кислой мальтазы, каждая из которых наследуется аутосомно-рецессивно. Биохимическая основа различных клинических проявлений этой ферментной недостаточности неясна. В младенческом возрасте недостаточность кислой мальтазы проявляется как общий гликогеноз. При рождении патологии не находят, но уже вскоре обнаруживают резкую мышечную слабость, кардиомегалию, гепатомегалию и заметное увеличение размеров языка. Накопление гликогена в моторных нейронах спинного мозга, а также в стволе мозга усугубляет мышечную слабость. Такие младенцы обычно умирают в течение первого года жизни. У детей и взрослых это заболевание проявляется как мышечная дистрофия. Детские формы заболевания характеризуются замедленным развитием ребенка, слабостью проксимальных мышц конечностей, увеличением размеров икроножных мышц. Заболевание может прогрессировать с развитием дыхательной недостаточности; смерть обычно наступает в конце 2-го десятилетия жизни. Может иметь место поражение сердца, однако гепатомегалия и макроглоссия встречаются редко. Заболевание у взрослых лиц начинается в 3—4-м десятилетиях жизни и ошибочно может быть диагностировано как конечностно-поясная дистрофия или полимиозит. Начальным проявлением заболевания служит дыхательная недостаточность, обусловленная слабостью диафрагмы. Печень, сердце и язык обычно не поражаются. Предположение о диагнозе возникает после исследования мышечного биоптата, в котором обнаруживают вакуоли, содержащие гликоген, и кислую фосфатазу. При электронной микроскопии видно, что гликоген как связан с мембранами, так и свободно располагается в тканях. Окончательный диагноз устанавливают при биохимическом исследовании пораженной мышцы. Активность кислой мальтазы в моче снижена. Уровень сывороточной активности КК может превышать норму в 10 раз. При ЭМГ можно дифференцировать мальтазную недостаточность от мышечной дистрофии по высокочастотным миотоническим разрядам, сопровождающим непродолжительные потенциалы моторных единиц, на фоне фибрилляций и положительных остроконечных потенциалов. Недостаточность фермента, тормозящего ветвление молекулы гликогена (гликогеноз III типа). Эта довольно легко протекающая детская болезнь проявляется гепатомегалией, замедлением роста и гипогликемией; нерезко выраженную мышечную слабость наблюдают редко. После пубертатного периода выраженность этих симптомов обычно уменьшается или они исчезают совсем, так что мышечная слабость и некоторое уменьшение мышечной массы могут быть связаны просто с уменьшением физической нагрузки из-за плохой к ней толерантности. Предположение о возможном диагнозе возникает тогда, когда после выполнения больным специального упражнения для мышц предплечья в крови не повышается содержание молочной кислоты. Сывороточная активность КК обычно повышена. При ЭМГ выявляют изменения, характерные для миопатии, а также признаки повышенной раздражимости мембран миотоническими импульсами. В биоптате мышцы обнаруживают вакуоли с повышенным содержанием гликогена. Для подтверждения диагноза требуется биохимическое исследование мышцы. Недостаточность гликоген-ветвящего фермента (гликогеноз IV типа). Недостаточность данного фермента — это очень тяжелая, фатальная патология младенческого возраста, при которой нарушения со стороны скелетной мускулатуры отходят на второй план по сравнению с развитием хронической печеночной недостаточности. Однако мышечная гипотония и атрофия мышц могут навести на мысль о первично мышечном заболевании или о спинально-мышечной атрофии. Недостаточность мышечной фосфорилазы (гликогеноз V типа). Плохая переносимость физической нагрузки является характерным симптомом недостаточности мышечной фосфорилазы, впервые описанной в 1951 г. Мак-Ардлем (McArdle). Заболевание наследуется аутосомно-рецессивно; мужчины болеют чаще, чем женщины. После пубертатного возраста у больных возникают болезненные мышечные судороги и быстрая утомляемость мышц после интенсивной физической нагрузки — бега, поднятия тяжестей. В литературе описаны варианты заболевания, начинающегося как в младенческом возрасте, так и позднее. Многие больные сообщают о феномене «второго дыхания», наступающего после кратковременного отдыха или после замедления темпа физической нагрузки, что позволяет им в течение долгих лет сохранять двигательную активность. Физическое переутомление у таких больных ведет к развитию рабдомиолиза, миоглобинурии и почечной недостаточности. Постоянная мышечная слабость и прогрессирующая атрофия мышц отмечаются редко, так что при физическом обследовании их в периоды между обострениями болезни патологии обычно не выявляют. Другие органы при данном заболевании не поражаются. Активность сывороточной КК подвержена значительным колебаниям и может быть повышена даже в бессимптомные периоды. Тест с нагрузкой на мышцы предплечья не сопровождается повышением в крови содержания молочной кислоты. Данные ЭМГ бывают нормальными, если только ее не осуществляют непосредственно после эпизода рабдомиолиза. В биоптате мышцы выявляют «пузырьки», содержащие гликоген под сарколеммой. Наличие недостаточности мышечной фосфорилазы может быть установлено с помощью гистохимической окраски гистологического препарата или при биохимическом исследовании мышечной ткани. Больные могут оставаться в течение жизни достаточно активными при условии воздержания от тех или иных физических перегрузок. Заместительная диетотерапия глюкозой или фруктозой обычно не сопровождается ослаблением симптоматики заболевания. Недостаточность фосфофруктокиназы (гликогеноз VII типа). Это заболевание напоминает недостаточность мышечной фосфорилазы и также наследуется по аутосомно-рецессивному типу; среди заболевших преобладают лица мужского пола. Те же, что и при фосфорилазной недостаточности, провоцирующие моменты и данные лабораторных исследований. Выявляют этот вид ферментной недостаточности при гистохимической окраске препарата мышцы на фосфофруктокиназу (ФФрК). Для достоверного диагноза необходимо биохимическое исследование мышечных ферментов. У некоторых больных с недостаточностью указанного фермента возможны нерезко выраженный гемолиз, увеличение числа ретикулоцитов в периферической крови, а также повышение содержания билирубина в крови, так как дефицит ФФрК имеет место при этом не только в мышцах, но и в эритроцитах. Синдромы, связанные с недостаточностью нового гликолитического фермента. Начиная с 1981 г. была идентифицирована недостаточность еще трех гликолитических ферментов: фосфоглицераткиназы (ФГлК) (тип IX), фосфоглицератмутазы (ФГлМ) (тип X) и лактатдегидрогеназы (ЛДГ) (тип XI). Клиническая картина всех этих трех типов ферментной недостаточности идентична. В раннем детстве или в подростковом возрасте после физических перенапряжений у больных возникают эпизоды миоглобинурии и миалгии. Думается, что все эти ферментные дефекты наследуются по аутосомно-рецессивному типу. Активность сывороточной КК может быть повышена как во время обострений заболеваний,
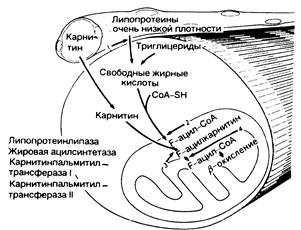
Рис. 357-1. Метаболизм липидов. Свободные жирные кислоты как энергетическое средство образуются из триглицеридов, накопленных в мышце, и из циркулирующих липопротеинов очень низкой плотности, которые расщепляются под влиянием эндотелиальной липопротеинлипазы (1) в капиллярах. Карнитин, эссенциальный субстрат для метаболизма липидов, образуется в печени и транспортируется к мышце. В мышце свободные жирные кислоты соединяются с коэнзимом A (CoA-SH) под влиянием жирной ацилсинтетазы (2), обнаруживаемой в наружной митохондриальной мембране, в результате чего образуется жировой ацилкоэнзим А (F-ацил-СоА). Транспорт через внутреннюю митохондриальную мембрану требует переноса карнитина с помощью карнитинпальмитинтрансфсразы I (КПТ-1), связанной с наружной поверхностью внутренней митохондриальной мембраны (3). Внутри митохондрии жировой ацилкарнитин (Р-ацилкарнитин) синтезируется с помощью КПТ-П (4), которая связана с внутренней поверхностью внутренней митохондральной мембраны. При этом жировой ацилкоэнзим А подвергается b-окислению.
так и между обострениями. При недостаточности ФГлМ и ЛДГ повышение содержания молочной кислоты в крови после нагрузки на мышцы предплечья обычно бывает ниже, чем в норме. При недостаточности ФГлК совсем не повышается содержание в крови лактата после нагрузки. И вообще, эта форма ферментной недостаточности по клиническим проявлениям очень напоминает недостаточность мышечной фосфорилазы и фосфофруктокиназы. Гистологическое исследование мышц при указанных формах энзимной недостаточности обычно неинформативно, отмечается лишь некоторое увеличение содержания в мышцах гликогена. Для достоверной диагностики необходимо биохимическое исследование мышцы. Нарушения метаболизма липидов. Липиды — это важный энергетический субстрат, особенно во время покоя мышцы и при длительных, но нерезких физических нагрузках (рис. 357-1). Недостаточность карнитина. Различают миопатическую и системную (генерализованная) формы карнитиновой недостаточности. Миопатическая карнитиновая недостаточность обычно протекает с генерализованной мышечной слабостью, которая, как правило, начинается в детстве. Клинические проявления этой болезни частично напоминают мышечную дистрофию, а частично — полимиозит. Большинство случаев спорадические; полагают, что болезнь может наследоваться по аутосомно-рецессивному типу. Иногда возникает кардиомиопатия. Активность сывороточной КК слегка повышена; на ЭМГ — признаки миопатии. В биоптате мышцы обнаруживают резко выраженное накопление липидов. Содержание карнитина в сыворотке крови нормальное. Считается, что при этом заболевании нарушается транспорт карнитина в мышцы, поэтому содержание его в мышцах столь низкое. Некоторые больные положительно отвечают на пероральную заместительную терапию карнитином, во всяком случае она должна быть испробована во всех случаях. Другие бальные по неизвестным причинам положительно отвечали на лечение преднизолоном. У некоторых больных лечебный эффект оказала замена в их диете триглицеридов со средней цепью на триглицериды с длинной цепью. Отдельные больные хорошо отвечают на лечение рибофлавином. Системная карнитиновая недостаточность — это аутосомно-рецессивное заболевание младенческого и раннего детского возраста. Оно характеризуется прогрессирующей мышечной слабостью и эпизодами печеночной энцефалопатии с тошнотой, рвотой, затемнением сознания, комой и ранней смертью. Низкое содержание карнитина в сыворотке крови отличает эту форму от миопатической карнитиновой недостаточности. Не установлено никакой причины, которая могла бы вызывать или объясняла низкое содержание карнитина в крови. У одних больных обнаруживают уменьшенный синтез карнитина, у других — повышенную его экскрецию с мочой. Активность сывороточной КК может быть слегка повышенной. В биоптате мышцы находят накопление липидов. В некоторых случаях их накопление отмечают также в печени, сердце и почках. У некоторых больных, но далеко не у всех, эффективным оказался пероральный прием карнитина или кортикостероидов. Карнитинпалмитилтрансферазная недостаточность. Указанная ферментная недостаточность проявляется повторяющейся миоглобинурией. Точно неизвестно, снижение активности какой карнитинпалмитинтрансферазы (КПТ) при этом происходит: КПТ-I или КПТ-II. Данная ферментная недостаточность, по-видимому, является результатом нарушения регуляции свойств патологического фермента. Большая физическая нагрузка (игра в футбол, длительный поход) может спровоцировать рабдомиолиз; однако иногда выявить провоцирующий фактор не удается. Первые признаки болезни часто появляются в детстве. В отличие от поражений мышц при нарушениях гликолиза, когда уже после кратковременных, но интенсивных физических нагрузок появляются судороги в мышцах, что заставляет больного отказываться от продолжения физической нагрузки и тем самым защитить себя, при КПТ-недостаточности боли в мышцах не возникают, пока не будут израсходованы все энергетические ресурсы мышцы и не начнется ее деструкция. Во время рабдомиолиза возникает резчайшая мышечная слабость, так что некоторым больным бывает необходима искусственная вентиляция легких. В отличие от карнитиновой недостаточности при недостаточности КПТ между приступами болезни мышечная сила сохранена, а при биопсии мышцы в ней не обнаруживают накопления липидов. Диагноз требует непосредственного исследования содержания в мышце КПТ. Лечение заключается в увеличении потребления углеводов с пищей перед физической нагрузкой или в замене в диете больного триглицеридов со средней цепью на триглицериды с длинной цепью. Однако все эти методы лечения не являются вполне удовлетворительными. Миоаденилатдеаминазная недостаточность. Фермент аденилатдеаминаза превращает 5-аденозинмонофосфат (5-АМФ) в инозинмонофосфат (ИМФ) с высвобождением аммиака, что может играть определенную роль в регулировании содержания в мышце аденозинтрифосфата (АТФ). В 1978 г. удалось выявить группу больных с мышечными болями и непереносимостью физической нагрузки, у которых имел место дефицит изоэнзима миоаденилатдеаминазы. Недостаточность этого фермента довольно распространена и встречается примерно у 1 % населения, что может быть установлено при специальной окраске мышечных гистологических препаратов или при биохимическом исследовании мышечной ткани. При исследовании теста с нагрузкой на мышцы предплечья обнаруживается снижение образования аммиака. Со времени оригинального описания данного заболевания более четких клинических его проявлений выявить не удалось. Нередко у больных с другой нервно-мышечной патологией (поражения клеток передних рогов спинного мозга, мышечная дистрофия, миастения) также обнаруживают недостаток этого фермента. Четко клиническое значение этого нарушения не установлено. Митохондриальные миопатии. Гетерогенная группа заболеваний, характеризующихся патологией со стороны митохондрий, обязана своим названием [ragged-red fibers («шероховатые красные волокна»)] особому виду окрашенного трихромно гистологического препарата биопсированной мышцы. Синдром Кирнса—Сейра — это спорадическое заболевание, начинающееся в детском возрасте и характеризующееся прогрессирующей экстернальной офтальмоплегией, нарушениями внутрисердечной проводимости, которая нередко приводит к полной поперечной блокаде. Отмечают также дегенерацию ретины, малый рост больных, гонадные дефекты. Наследственное заболевание с прогрессирующей экстернальной офтальмоплегией и слабостью проксимальных мышц бывает трудно отличить от синдрома Кирнса— Сейра. Недавно был выделен еще один синдром, обозначаемый акронимом MERRF1, при котором миоклоническая форма эпилепсии сочетается с обнаруживаемыми в гистологических мышечных препаратах «шероховатыми красными волокнами». Заболевание это возникает между 1-ми 5-м десятилетиями жизни и характеризуется генерализованными судорожными припадками, миоклонусом, деменцией, потерей слуха и атаксией. Третье заболевание из этой группы — это МЕLАS2-синдром (1 MERRF — myotonic epilepsy, ragged-red fibers (примеч. ред.). 2 MELAS — myopathy encephalopathy, lactic acidosis, stroke-like episodes (примеч. ред.)., который является медленно прогрессирующим заболеванием, характеризующимся митохондриальной миопатией, энцефалопатией, молочнокислым ацидозом, инсультоподобными эпизодами с развитием преходящих гемипарезов, гемианопсией или кортикальной слепотой, а также очаговыми или генерализованными судорожными припадками. Причина митохондриальных миопатий неизвестна, однако имеются факты в пользу того, что в семейных случаях болезнь может передаваться митохондральной, а не хромосомной ДНК.
Воспалительные миопатии
Полимиозит и дерматомиозит (см. гл. 356) обычно развиваются медленно, в течение месяцев. Наличие характерных кожных изменений при дерматомиозите значительно облегчает диагностику. Хронические формы полимиозита с медленно прогрессирующей слабостью проксимальных мышц часто бывает невозможно отдифференцировать на основании клинической картины от спорадических случаев поясно-конечностной дистрофии. В таких случаях не всегда помогают и детально отснятая ЭМГ, и даже биопсия мышцы. Подострая или хроническая воспалительная миопатия была выделена в особую подгруппу и названа миозитом с вирусными включениями. В названии отражено наличие в цитоплазматических мембранах и в ядрах особых включений, состоящих из патологических филаментов. Болезнь не поддается лечению кортикостероидами. Хронический миозит встречается также при всех коллагеново-васкулярных заболеваниях, а также при саркоидозе.
Эндокринные и метаболические миопатии
Многим эндокринным заболеваниям сопутствует мышечная слабость. Причина этого далеко не всегда ясна. При этом даже не бывает понятно, связано это с собственно мышцей или с какой-то другой частью моторной единицы, поскольку активность сывороточной КК при этом остается нормальной, а в биоптате мышцы обнаруживают скорее атрофию, нежели деструкцию мышечных волокон. Названные миопатии хорошо «отвечают» на адекватное лечение соответствующего эндокринного заболевания. Патология щитовидной железы (см. гл. 324). Иногда гипертиреоз может дебютировать мышечной слабостью. Следует отметить, что как гипер-, так и гипотиреоз могут сопровождаться мышечной слабостью и болями в мышцах. Активность сывороточной КК часто бывает повышена, иногда почти в 100 раз превышая норму даже при минимальных признаках поражения мышц. У взрослых больных может отмечаться мышечная гипертрофия с судорогами (синдром Гоффманна), а у детей встречается сочетание кретинизма с отчетливой миопатией, сопровождающейся мышечной гипертрофией (синдром Kocher — Debre — Semelaigne). Патология паращитовидных желез (см. гл. 336). Гиперпаратиреоз часто сопровождается мышечной слабостью и атрофией мышц, нередко с «мышечными» болями, на самом деле имеющими скорее костное происхождение. Гиперпаратиреоз нередко проявляется неврологическими симптомами. Нервно-мышечные проявления характеризуются тетанией, но так как активность сывороточной КК бывает повышена, то заболевание обычно принимается за полимиозит. Типичны гиперрефлексия или арефлексия, несмотря на наличие признаков Хвостека и Труссо. Патология надпочечников (см. гл. 325). Эндогенное повышение в крови уровня кортикостероидов может вызывать резкую мышечную слабость и уменьшение мышечной массы. Недостаточность надпочечников часто сопровождается вялостью мышц и их слабостью, несмотря на сохранение мышечной силы при объективном исследовании. Патология гипофиза (см. гл. 321). В отдельных случаях акромегалия сопровождается увеличением мышц. Встречается и мышечная слабость миопатического типа, однако слабость эта скорее является результатом сопутствующей эндокринной патологии или невропатии. Мышечная же слабость при панпитуитаризме, вероятно, возникает вследствие одновременно существующих недостаточности надпочечников и щитовидной железы. Сахарный диабет (см. гл. 327). Слабость проксимальных мышц у больных сахарным диабетом обычно является результатом невропатии. Обнаружение же при ЭМГ или биопсии мышцы признаков миопатии, а также повышение активности сывороточной КК обычно указывают на какое-то сопутствующее заболевание. Витаминная недостаточность. Синдром мальабсорбции, особенно в раннем детстве, может привести к миопатии, связанной с недостаточностью витамина Е. При миопатиях другого генеза витамин Е обычно не применяют для лечения больных (см. гл. 76). Дефицит же витамина D (см. гл. 337) независимо от того, возникает ли он в связи с его сниженным употреблением, с нарушением его абсорбции или обусловлен патологией почек, может повести к хронической мышечной слабости; болевые ощущения при этом скорее отражают патологию костей. Недостаток других витаминов обычно не вызывает миопатии. Другие метаболические расстройства. Такие системные заболевания, как злокачественные новообразования, хроническая дыхательная, сердечная, печеночная или почечная недостаточность, часто сопровождаются существенным уменьшением мышечной массы и жалобами на мышечную слабость. При этом тесты на мышечную силу остаются удивительно близкими к норме, так что жалобы больных можно объяснить плохой переносимостью мышечного напряжения. Признаков мышечного заболевания, как правило, нет. Мышечная слабость в таких случаях может быть обусловлена нарушениями в электролитном обмене, в частности хроническими гипокалиемией, гиперкальциемией или гипокальциемией различной этиологии.
Токсические миопатии
Классификация токсических миопатий приведена в табл. 357-2.
Таблица 357-2. Токсические миопатии
1. Фокальные миопатии: пентазоцин, меперидин 2. Генерализованные миопатии а) воспалительные: циметидин, D-пеницилламин, новокаинамид; б) мышечная слабость и миалгии: хлорохин, клофибрат, колхицин, кортикостероиды, эметин (Emetine), эпсилон-аминокапроновая кислота, лабеталол, пергексилен (Perhexilene), анаприлин (пропранолол), винкристин; в) рабдомиолиз и миоглобинурия: алкоголь, азатиоприн, героин, амфетамин, клофибрат, эпсилон-аминокапроновая кислота, фенциклидин (Phencyclidine), барбитураты; г) злокачественная гипертермия: фторотан. этилен, диэтиловый эфир, метоксифлуран, этил-хлорид (Ethyl chloride), трихлорэтилен, галламин (Gallamine), дитилин, лидокаин, меливакаин (Mepivacaine)
Лекарственные препараты и различные химические агенты могут вызывать как местное, так и общее повреждение скелетной мускулатуры. Наиболее частой причиной местного поражения мышц являются инъекции наркотических анальгетиков. В этом отношении следует обратить внимание на два препарата [пентазоцин и меперидин (Meperidine)], которые могут вызывать резко выраженную фибротическую реакцию со стороны мышц. Инъекции обычно делают в дельтовидную, трехглавую (трицепс), большую ягодичную и четырехглавую мышцы. При этом мышцы становятся твердыми, в них нередко формируются абсцессы. Кожа над ними нередко изъязвляется и западает. Иногда появляются резко выраженные контрактуры суставов. Другие препараты могут вызывать генерализованную мышечную слабость, что в большей степени проявляется в проксимальных мышцах. В большинстве случаев точный механизм лекарственной токсичности неизвестен. Так. D-пеницилламин вызывает состояние, симулирующее дерматомиозит и полимиозит. Были сообщения об аналогичном состоянии, возникшем после употребления циметидина. Новокаинамид может вызвать миозит как часть волчаночноподобной реакции. Хлорохин после многомесячного применения приводит к четко выраженной вакуолярной миопатии, иногда с поражением миокарда. Клофибрат вызывает слабость и боли в мышцах, иногда вскоре после начала лечения, а иногда спустя несколько месяцев. Повышение активности сывороточной КК может быть при этом единственным проявлением отрицательного влияния этого препарата на мышцы. Слабость в мышцах и некроз мышечных волокон могут развиться после нескольких недель лечения эметином гидрохлоридом (используемым при лечении амебиаза), эпсилон-аминокапроновой кислотой (антифибролитический агент) и пергексиленом (применяемым при грудной жабе). Лекарственная миопатия возникает и при лечении кортикостероидами, при этом весьма характерна слабость проксимальной мускулатуры. Мышечную слабость особенно склонны вызывать кортикостероиды с наличием фтора в 9a-позиции; это — триамцинолон, дексаметазон и бетаметазон. Практически же длительное применение всех кортикостероидов, включая преднизон, приводит к развитию мышечной слабости. Прием указанных препаратов несколько раз в день вызывает большую мышечную слабость, чем однократный их прием утром. Одноразовое в течение дня их применение или прием этой дозы через день в большей мере щадит мышечную систему (см. гл. 325). Клинический диагноз стероидной мышечной слабости может быть очень труден, если кортикостероиды применяются по поводу воспалительной миопатии. В подобных ситуациях в пользу вызванной кортикостероидами мышечной слабости свидетельствуют нормальная активность сывороточной КК, нормальная (или с минимальными миопатическими отклонениями) ЭМГ и наличие в мышечном биоптате атрофии мышечных волокон II типа. В некоторых случаях токсическая миопатия может носить катастрофический характер, вызывая рабдомиолиз и миоглобинурию (см. гл. 376). Очень серьезным осложнением медикаментозной терапии является злокачественная гипертермия (см. гл. 8), которая возникает у лиц, к ней предрасположенных, после применения общих анестетиков и деполяризующих мышечных релаксантов (см. табл. 357-2). Используемые при местной анестезии лидокаин и мепивакаин иногда также могут спровоцировать названное осложнение.
Дата добавления: 2015-02-02 | Просмотры: 1450 | Нарушение авторских прав |